April 26, 2024
Written by Marie Diamond and Maria Rizzo Systematic literature reviews (SLR) are essential to informing healthcare...
Read article
April 22, 2024
Written by Boaz N. Adler, Director, Global Product Engagement, and J. Kyle Wathen, Vice President, Scientific Strategy...
Read article
April 17, 2024
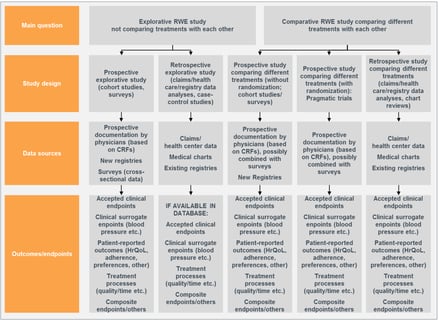
Written by Grace Hsu, Evie Merinopoulou, and Jason Simeone To establish treatment efficacy and safety, regulatory and...
Read article
April 15, 2024
In the context of clinical trials, reducing the workload of the clinical team without compromising data quality is...
Read article
April 12, 2024
How should health technology developers prepare for future market access activities in Europe? Numerous discussions...
Read article
April 10, 2024
Orphan drug designation is a regulatory status granted to pharmaceuticals developed for the treatment of rare diseases....
Read article
April 8, 2024
In this latest edition of the Career Perspectives series, we are excited to introduce our readers to Joshua Murray,...
Read article
April 5, 2024
Written by Sydney Ringold, Customer Success Manager, and Kevin Trimm, Chief Product Officer In an ever-changing...
Read article
April 3, 2024
Thank you to Charlotta Gauffin, Chief Scientific Officer at Dicot, for joining us for our recent webinar, “The Road to...
Read article
April 2, 2024
Data monitoring committees (DMCs) review ongoing clinical trial data to make recommendations regarding trial conduct...
Read article
March 20, 2024
Written by Lydia Vinals, PhD, and Grammati Sarri, PhD The draft Implementing Act of the EU Health Technology Assessment...
Read article
March 13, 2024
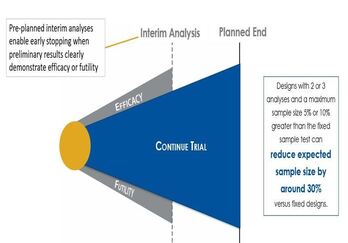
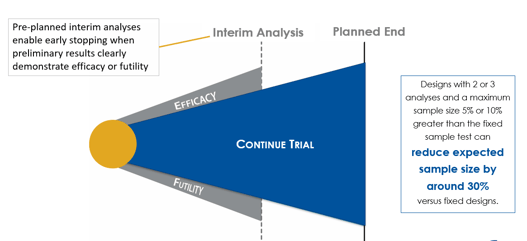
Group sequential clinical trial designs — a type of adaptive clinical trial design — have emerged as a powerful tool in...
Read article
March 11, 2024
In 2023, rare diseases accounted for 30% of product pipeline under development, about half of which comprising...
Read article
March 4, 2024
In this new installment of the Career Perspectives series, we had the privilege of interviewing Arnold van Aswegen,...
Read article
February 26, 2024
Written by Boaz N. Adler, MPA, Director, Global Product Engagement, and Valeria Mazzanti, MPH, Associate Director,...
Read article
February 20, 2024
In clinical trials based on count data, the aim is to compare independent treatment groups in terms of the rate of...
Read article
February 14, 2024
During drug development, a comprehensive regulatory strategy is key for saving time and money. There are many common...
Read article
February 13, 2024
Written by Boaz N. Adler, MPA, Director, Global Product Engagement, and Valeria Mazzanti, MPH, Associate Director,...
Read article
February 2, 2024
When building a disease model or an economic model, the assumption has been that updates to such models should only...
Read article
January 26, 2024
Innovations in the process of designing adaptive clinical trials have unlocked new possibilities for designing and...
Read article
January 22, 2024
Regulatory guidelines outline all crucial studies and documentation that should be in place before a drug product can...
Read article
January 19, 2024
Independent data monitoring committees help to ensure patient safety and uphold trial integrity. In Central Nervous...
Read article
January 17, 2024
Despite accumulating learnings from early phases, several uncertainties remain to be addressed when designing pivotal...
Read article
January 15, 2024
After explosive and frenetic activity in the clinical trial industry during the COVID era, the past two years have seen...
Read article
January 10, 2024
A clinical development strategy is a comprehensive plan designed to establish the safety and efficacy of new...
Read article
January 3, 2024
Ten years ago this month, in January 2014, the FDA issued the first version of its Technical Conformance Guide (by...
Read article
December 29, 2023
What a year! Perspectives has explored a myriad of topics this year within clinical development — from adaptive trial...
Read article
December 27, 2023
Perspectives covers a wide range of topics related to data submission and data integration, from ISS and ISE best...
Read article
December 22, 2023
Perspectives covers a wide range of topics within therapeutics development from advice on regulatory submission to...
Read article
December 20, 2023
Perspectives covers a wide range of topics related to real-world evidence and real-world data, from overcoming health...
Read article
December 15, 2023
In clinical trials, high-quality data is essential. It drives the drug development decision-making process and is a...
Read article
December 8, 2023
Regular technical discussions with the FDA play a critical role in ensuring data submission success. These discussions...
Read article
December 6, 2023
Chemistry, Manufacturing, and Controls (CMC) is a critical component of drug product development. As a Senior...
Read article
December 4, 2023
Independent data monitoring committees review unblinded clinical trial data and issue recommendations to designated...
Read article
December 1, 2023
Last week, Robert Szulkin, Research Principal, Real World Evidence, discussed the need for real-world evidence studies,...
Read article
November 29, 2023
A clinical trial is usually performed using some kind of comparator. This could be another drug on the market, or a...
Read article
November 24, 2023
Regulatory requirements regarding documentation for new medicines are constantly evolving. Previously, randomized...
Read article
November 20, 2023
Interpreting all guidelines before your first-in-human clinical trials can be overwhelming. While guidelines are...
Read article
November 17, 2023
Cytel’s Functional Service Provision (FSP) teams work on exciting projects with biotech and pharmaceutical companies as...
Read article
November 13, 2023
For nonclinical studies that precede Phase I, a drug formulation in high doses and concentrations is required. While...
Read article
November 10, 2023
Independent data monitoring committees review unblinded clinical trial data and issue recommendations to designated...
Read article
November 8, 2023
Written by Natalia Muehlemann, Vice President, Clinical Development, and Ari Brettman, Senior Managing Director,...
Read article
November 6, 2023
The first-in-human trial, which aims to show the safety and tolerability of a new drug, is a major milestone for any...
Read article
November 3, 2023
Written by Fei Tang and Evie Merinopoulou Reinforcement Learning (RL), a crucial component of machine learning (ML),...
Read article
November 1, 2023
It feels like just yesterday I attended my first PHUSE conference back in 2005 in Heidelberg, Germany. Fast forward 19...
Read article
October 30, 2023
The past two decades have seen the adoption of great innovation in clinical trial design. Statisticians have risen to...
Read article
October 27, 2023
Written by Natalia Muehlemann, Vice President, Clinical Development; Martin Frenzel, Research Principal, Statistical...
Read article
October 25, 2023
The European Union (EU) health technology assessment (HTA) regulation aims to improve the availability of innovative...
Read article
October 23, 2023
The Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER) are the...
Read article
October 20, 2023
Written by Grammati Sarri and Yannis Jemiai The spotlight for this year’s World Evidence-Based Healthcare Day (EBHC)...
Read article
October 18, 2023
Written by Angelo Tinazzi and Florence Le Maulf Integrated Summaries of Safety (ISS) and Integrated Summaries of...
Read article
October 13, 2023
Written by Alind Gupta, Cytel; Haridarshan Patel, Horizon Therapeutics; and Jason Simeone, Cytel Ophthalmology is...
Read article
October 11, 2023
Moving beyond static evidence development to ensure local market access success; responding to recent changes in...
Read article
October 10, 2023
Cytel’s Functional Service Provision (FSP) teams work on exciting projects with biotech and pharmaceutical companies as...
Read article
October 6, 2023
Real-world evidence studies are becoming increasingly popular in pharmaceutical development. But to ensure such studies...
Read article
October 4, 2023
Adaptive clinical trial designs have become increasingly popular among developers and investors due to the many...
Read article
October 2, 2023
Patients’ adherence to the medications or treatment regimens prescribed to them by their clinicians is an important...
Read article
September 29, 2023
In the ever-changing field of clinical trial design, there is often a need to evaluate design options quickly and...
Read article
September 25, 2023
As the pressure to reduce timelines rises across the industry, independent data monitoring committees (IDMCs) — which...
Read article
September 20, 2023
Cytel’s Functional Service Provision (FSP) teams work on exciting projects with biotech and pharmaceutical companies as...
Read article
September 18, 2023
With the ongoing changes inherent in the life sciences industry comes heightened expectations for companies to take...
Read article
September 13, 2023
Independent data monitoring committees (IDMCs) review ongoing clinical trial data to make recommendations regarding...
Read article
September 11, 2023
As a drug developer, you have to live with the answers and comments you get from regulatory authorities. Therefore,...
Read article
September 8, 2023
Single-arm trials, unlike placebo-controlled randomized control trials, forgo the use of a placebo or standard-of-care...
Read article
September 1, 2023
Data is the cornerstone of any clinical trial, driving the decision-making process of drug development, and is a...
Read article
August 30, 2023
Cytel’s Functional Service Provision (FSP) teams work on exciting projects with biotech and pharmaceutical companies as...
Read article
August 28, 2023
A clinical development plan — a comprehensive strategy for developing an investigational product through regulatory...
Read article
August 25, 2023
For several years, CDISC and Regulatory Data Submission expert Angelo Tinazzi has authored the series, The Good Data...
Read article
August 22, 2023
Written by Cyrus Mehta and Heather Struntz The significant time and cost, as well as high failure rates, of clinical...
Read article
August 18, 2023
The most common cause for incomplete Phase III trials is enrollment. Indeed, as many as 37% of trials miss...
Read article
August 14, 2023
In the first part of this post, I discussed the ongoing revolution, or maybe I should say evolution, we are living...
Read article
August 11, 2023
Systematic literature reviews are essential for proving product value to health authorities, clinicians, and payers,...
Read article
August 9, 2023
How do you envision the future of data submission? Last week, I had the privilege of presenting the topic “Standards...
Read article
August 4, 2023
“There is always the risk that interim analyses might occur after the Sufficient Information Threshold has been...
Read article
August 1, 2023
Evaluating the efficacy and safety of novel therapies in rare indications can be challenging due to the difficulty of...
Read article
July 28, 2023
Written by Fei Tang, RWE Senior Research Consultant, and Paul Arora, Assistant Professor (Status), Dalla Lana School of...
Read article
July 14, 2023
Written by Grammati Sarri, David Smalbrugge, Andreas Freitag, and Evie Merinopoulou The vision of a single, centralized...
Read article
June 20, 2023
Legislation on pediatric studies has existed for more than 20 years in the US, yet additional guidance from the FDA has...
Read article
June 14, 2023
Medical research has come a long way in recent years, fueled by innovative trial designs that challenge traditional...
Read article
June 9, 2023
In the ever-evolving landscape of clinical development, the need for robust evaluation of interim clinical data through...
Read article
June 7, 2023
Since its founding in 1987, Cytel has been home to some of the most passionate, creative, and talented experts in the...
Read article
June 5, 2023
Cytel recently announced the launch of its Therapeutics Development Team, bringing together quantitative,...
Read article
June 2, 2023
Data submissions are very regulated, but every drug and drug development are different. Therefore, the data presented...
Read article
May 24, 2023
As of March 2023, specifically for any study started on or after March 15, 2023,1 for the submission of SEND, SDTM, and...
Read article
May 22, 2023
By Grammati Sarri, Evie Merinopoulou, Vinusha Kalatharan, and Jason Simeone The Canadian Agency for Drugs and...
Read article
May 10, 2023
The evidence is staggering on the unequal health burdens experienced by specific patient groups defined by ethnic,...
Read article
May 5, 2023
As clinical trials become more complex, simulation-guided design approaches are crucial. For this edition of the...
Read article
May 3, 2023
With input by Alind Gupta, Louis Dron, and Jason Simeone. Randomized clinical trials (RCTs) have long been considered...
Read article
April 26, 2023
Regular technical discussions with the FDA play a critical role in ensuring data submission success. These discussions...
Read article
April 25, 2023
An interview with Miguel Hernán, Harvard University Kolokotrones Professor of Biostatistics and Epidemiology On March...
Read article
April 24, 2023
In the last 10 years, the Asia-Pacific (APAC) region has become a hotspot for clinical trials: the region contributed...
Read article
April 21, 2023
It was early March 2020, after the world was hit by the Covid-19 pandemic, that those of us on the CDISC Eu committee...
Read article
April 18, 2023
The Inflation Reduction Act (IRA), passed in August 2022, marks a significant shift in the US healthcare landscape,...
Read article
April 14, 2023
Real-world data and evidence are increasingly being used in health care decisions and publications. However, there are...
Read article
April 5, 2023
Health technology assessment (HTA) submissions require cost effectiveness analyses based on comparative effectiveness...
Read article
March 24, 2023
Last week, we featured our final Winter Weekend Read, the last in a series designed to showcase our complimentary...
Read article
March 21, 2023
One of the lesser-known complications associated with Multiple Sclerosis is a higher risk of serious infections (SIs)....
Read article
March 17, 2023
Smaller clinical trials can be optimized in significant ways using simulation-guided design. A small biotech studying...
Read article
March 13, 2023
Many industries have long since adopted the practice of modeling and simulating experimental scenarios. And despite...
Read article
March 8, 2023
Real-world data has been increasingly used to answer questions related to the course, prognosis, and treatment of...
Read article
March 7, 2023
Health inequalities are an enduring issue that can be exacerbated by clinical trial recruitment that does not reflect...
Read article
March 3, 2023
The speed of scientific discovery has been outpacing the ability of researchers to accumulate and integrate constantly...
Read article
February 24, 2023
Last week, Cytel Director & Research Principal Louis Dron discussed new FDA guidance on the design and conduct of...
Read article
February 22, 2023
Simulation-guided design is quickly becoming a novel feature of modern drug development. Its foundational promise is to...
Read article
February 14, 2023
The U.S. FDA has recently provided specific guidance[i] on the design and conduct of trials incorporating an external...
Read article
February 9, 2023
Those familiar with simulation-guided design (SGD) know that it can be used for a wealth of clinical trial options:...
Read article
February 8, 2023
Over the past years, probably the entire last decade, there have been several discussions on how to handle multiple...
Read article
February 3, 2023
For clinical development and research and development teams, the Pareto Frontier can perform two functions. Let’s take...
Read article
January 31, 2023
Asthma affects more than 235 million people worldwide, and due to lacking effective implementation of clinical...
Read article
January 27, 2023
Should your clinical trial be adaptive? Trials that include a prospectively planned modification based on an interim...
Read article
January 25, 2023
People think in Bayesian terms all the time: we use prior information and the evidence at hand to make decisions in our...
Read article
January 23, 2023
Ever since the first immune checkpoint inhibitor was approved for market nearly twelve years ago, the industry has...
Read article
January 20, 2023
A small biotech’s conventional carcinoma trial design was too expensive to implement and did not offer the opportunity...
Read article
January 17, 2023
When it comes to rare diseases, a handful of major challenges to drug development arise. Bayesians strategies have...
Read article
January 13, 2023
Bayesian methods, with their ability to facilitate flexibility and learning, are often associated with early-phase...
Read article
January 12, 2023
Statistical methods have long been fundamental to drug development, and advancements in the last few decades in...
Read article
January 6, 2023
You may have heard that our clinical trial strategy platform Solara® won the Fierce Life Sciences award for Technology...
Read article
January 4, 2023
Returning to Cytel after the winter holidays, I am excited to begin a year that will likely prove memorable for both my...
Read article
December 27, 2022
Bayesian methods have been playing a key role in transforming clinical research, providing a variety of new...
Read article
December 21, 2022
Perspectives on Enquiry and Evidence features two recurring interview series: Our new Industry Voices series, in which...
Read article
December 19, 2022
Adaptive trial designs – that is, trials that include a prospectively planned modification based on an interim analysis...
Read article
December 16, 2022
Bayesian methods have been playing a key role in transforming clinical research, and Bayesian topics are frequently...
Read article
December 9, 2022
As promised in my last post prior to PHUSE-EU Connect, I’d like to now share some reflections on my “Integration...
Read article
December 6, 2022
In this edition of the Career Perspectives series, I spoke with Veronica Chan, Principal Clinical Data Manager at...
Read article
December 2, 2022
For 35 years, Cytel’s scientific rigor and operational excellence have enabled biotech and pharmaceutical companies to...
Read article
November 29, 2022
Bayesian methods have continuously played a key role in transforming clinical research in therapeutic areas such as...
Read article
November 18, 2022
In the following interview, Dr. Parvin Fardipour, Quantitative Strategies & Data Science, sits down with Heather...
Read article
November 14, 2022
When conducting network meta-analysis (NMA) – that is, a technique that involves comparing multiple treatments...
Read article
November 9, 2022
In this edition of the Career Perspectives series, I interview Malte Stein, Senior Biostatistician. Malte owned the...
Read article
November 8, 2022
Although many of you can’t wait for the start of the Football World Cup 2022 (less than two weeks while I’m writing),...
Read article
November 3, 2022
Cytel will be represented at over 60 presentations at ISPOR Europe 2022, with more issue panels and workshops than any...
Read article
November 2, 2022
Pharmaceutical research in oncology is increasingly focused on the development of therapies targeted at newly...
Read article
October 28, 2022
Integrating the “pressure testing” of clinical trial designs into the process of creating a strong clinical trial...
Read article
October 24, 2022
On the occasion of Cytel’s 35th anniversary, co-founder Professor Cyrus Mehta sits down with Dr. Esha Senchaudhuri to...
Read article
October 18, 2022
On the occasion of Cytel’s 35th anniversary, co-founder Professor Nitin Patel sits down with Dr. Esha Senchaudhuri to...
Read article
October 14, 2022
In this edition of the Career Perspectives series, I interview Nicolas Rouillé, Senior Director, Statistical...
Read article
October 13, 2022
ISPOR Europe, the leading global conference for health economics and outcomes research (HEOR) and real-world evidence...
Read article
October 11, 2022
On the occasion of Cytel’s 35th anniversary, our CEO Joshua Schultz sits down with Dr. Esha Senchaudhuri to discuss the...
Read article
October 4, 2022
Integrated Nested Laplacian Approximations (or INLA) are now starting to be used by statisticians as a key tool for...
Read article
October 3, 2022
The wider availability of electronic health data, medical registries, and even larger proprietary datasets means that...
Read article
September 28, 2022
The landscape of drug development has changed dramatically over the last few decades, and effective statistical leaders...
Read article
September 27, 2022
On Louis Dron et al., “Data Capture and Sharing in the COVID-19 Pandemic: A Cause for Concern,” The Lancet 4 (10) (2022)
Read article
September 23, 2022
Adaptive trial designs – that is, trials that include a prospectively planned modification based on an interim analysis...
Read article
September 16, 2022
In the first part of this article, I raised awareness of the availability of additional FDA guidances containing CDISC...
Read article
September 15, 2022
On August 16, 2022, President Biden signed into law the Inflation Reduction Act of 2022, which includes U.S. drug...
Read article
September 12, 2022
In clinical trials, patient enrollment is often staggered, with data collected sequentially. When designing a clinical...
Read article
September 9, 2022
A new trend has emerged over the last decade that has changed the way many clinical trials are conducted. Unlike...
Read article
September 8, 2022
The American Statistical Association Biopharmaceutical Section, in cooperation with the FDA Statistical Association,...
Read article
August 26, 2022
To continue our Summer Weekend Reads series, Cytel presents “Discover the Value of an Optimized Clinical Data Strategy”...
Read article
August 25, 2022
The International Society for Pharmacoepidemiology is hosting its “ICPE 2022: Advancing Pharmacoepidemiology and...
Read article
August 18, 2022
The American Statistical Association’s annual Joint Statistical Meeting (JSM) gathered over 6,500 attendees from 52...
Read article
August 4, 2022
In this edition of the Career Perspectives series, I interview Allison Luccock, Director of Business Operations for...
Read article
August 3, 2022
A number of late-phase clinical trial sponsors remain hesitant to employ Bayesian approaches in confirmatory settings,...
Read article
July 28, 2022
When new treatments are compared with existing therapies in clinical care, population-adjustment techniques need to...
Read article
July 27, 2022
Written by Jing Ping Yeo and Charles Warne Adaptive designs are studies that “include a prospectively planned...
Read article
July 26, 2022
To watch this webinar and others from this introductory series, click the link below. The ability to draw on electronic...
Read article
July 20, 2022
When performing indirect treatment comparisons, effect modification can create complexities in the event of high...
Read article
July 13, 2022
Many thanks to Grammati Sarri and Michael Groff for their comments in developing this blog. An indirect treatment...
Read article
July 7, 2022
The gold standard for assessing the efficacy for a medicine continues to be RCTs, however, for many reasons (disease...
Read article
June 28, 2022
Founded in 1987 by Cyrus Mehta and Nitin Patel, research scientists at Harvard University and MIT respectively and...
Read article
June 15, 2022
For years CDISC data standards implementers have struggled to find good implementation examples and use cases beside...
Read article
June 14, 2022
Suppose you had to choose six clinical trials intended for registration with regulatory agencies, only six, to explain...
Read article
June 13, 2022
The International COVID-19 Data Alliance (ICODA) was formed to address the challenge of generating rapid and rigorous...
Read article
June 8, 2022
Clinical researchers, seeking to understand the statistical benefits of a common Phase 2 oncology design, now have a...
Read article
June 7, 2022
In most instances of blinded sample size re-estimation, the timing of the interim analysis that determines whether the...
Read article
June 2, 2022
How can clinicians at the forefront of modern clinical trials and statisticians at the forefront of advanced...
Read article
May 27, 2022
When submitting systematic literature reviews to a Health Technology Assessment authority, high volumes of research...
Read article
May 26, 2022
When constructing estimands a key question that arises is how to handle intercurrent events and missing data. In a...
Read article
May 24, 2022
Traditionally, clinical trials are expensive, long in duration, and have low success rate. But with the advent of rich...
Read article
May 16, 2022
A combination of industry and policy forces have recently changed the shape of Australia’s R&D sector, making it a...
Read article
May 12, 2022
Synthetic control arm (SCA) methods are statistical methods that are seeing rapidly increasing use in comparative...
Read article
May 5, 2022
While randomized control trials remain the industry gold-standard for regulatory and reimbursement submissions, there...
Read article
May 4, 2022
If you are a pharmaceutical or biotech company seeking to enter the market with a new drug, you need to submit a...
Read article
May 3, 2022
In 2005, Pfizer launched a Phase 1 trial for the kinase inhibitor crizotinib. Six years later, it was approved, thanks...
Read article
April 29, 2022
At ISPOR US 2022, Cytel’s HEOR & RWE experts will be contributing to a range of Issue Panels, In-person Podium...
Read article
April 27, 2022
New medicines and devices under development live and die on the strength of their clinical data. An asset’s journey is...
Read article
April 25, 2022
I am excited to see you all at the CDISC Europe Interchange, April 27 – 28 but unfortunately, it will be a virtual...
Read article
April 22, 2022
This past decade has undoubtedly witnessed an increase in the number of single arm trials submitted to HTA bodies....
Read article
April 21, 2022
Sophisticated Bayesian Methods are gaining a lot of traction as they bring flexibility and speed to clinical trial...
Read article
April 13, 2022
The Life Sciences landscape has seen an impactful digital evolution in the past two years. The pandemic has accelerated...
Read article
April 12, 2022
In this edition of the Career Perspectives series, I interview Charles Warne, Associate Director of Biostatistics at...
Read article
April 8, 2022
The planning and optimization of a clinical trial is beset by uncertainties: knowledge of treatment effects, the...
Read article
April 7, 2022
Randomized control trials (RCTs) are the gold standard for estimating the efficacy of a treatment. They allow us to...
Read article
April 5, 2022
Hello! I’m delighted to pen my inaugural blog post here as Cytel’s Chief Medical Officer. In this series, we’ll explore...
Read article
April 1, 2022
At the end of every year, scientists from across Cytel’s business units are nominated for Cytel’s Spotlight Awards,...
Read article
March 23, 2022
For over a decade, the number one reason cited for trial discontinuity has been challenges associated with recruitment...
Read article
March 18, 2022
Real world evidence (RWE) provides a large and growing source of insights into drug uptake and safety. It is...
Read article
March 11, 2022
Every year, sponsors hesitating to use a complex innovative clinical trial design routinely miss opportunities to...
Read article
March 8, 2022
The issue of delayed treatment effects in immuno-oncology was demonstrated during a FDA-Industry sponsored workshop...
Read article
February 22, 2022
When determining the best possible statistical design for a particular trial, large pharmaceuticals and small biotechs...
Read article
February 18, 2022
Data is the most crucial asset of any clinical trial and hence, sponsors cannot jeopardize collecting clean...
Read article
February 17, 2022
It is a common perception that the role of a Data Manager is only to perform what we call “Data Cleaning”; making sure...
Read article
February 15, 2022
As the use of advanced and innovative clinical trial designs continue to rise, sponsors often wonder which estimation...
Read article
February 11, 2022
Adopting data standards such as CDISC in the early phase of clinical drug development contributes to the consolidation...
Read article
February 9, 2022
Data is the cornerstone of any clinical trial and is used to ultimately drive the decision-making process related to...
Read article
February 4, 2022
The ability to conduct data-driven and quantitatively rigorous feasibility studies, is often key to successful trial...
Read article
February 2, 2022
For nearly ten years, suboptimal trial enrollment has been cited as a primary cause of clinical trial discontinuation....
Read article
January 27, 2022
As a part of Cytel’s 10 Year Anniversary of the Promising Zone Design, Cytel hosted a quiz on “Keeping the Promise” –...
Read article
January 26, 2022
In this edition of the Career Perspectives series, I interview Jessica Bhoyroo, Cytel Clinical Data Manager based in...
Read article
January 25, 2022
In this blog, I share some experiences we recently had during an FDA submission Cytel performed for a sponsor after...
Read article
January 20, 2022
Limited patient populations resulting in small study sample sizes is a difficulty associated with the development of...
Read article
January 19, 2022
The past two years have been transformative for Cytel. Most notably, the global COVID-19 pandemic unleashed an...
Read article
December 23, 2021
Cytel blogs bring you debate and discussion of the newest trends in statistics and quantitative strategy. In 2021, our...
Read article
December 20, 2021
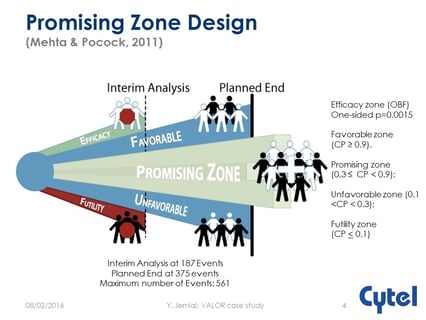
The promising zone design is an adaptive design which allows for sample size re-estimation based on the results of an...
Read article
December 15, 2021
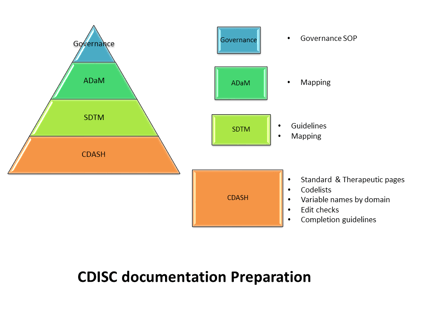
Recently, on November 29 I received an email from CDISC announcing an important update for both SDTM and ADaM CDISC...
Read article
December 14, 2021
In traditional clinical trial design, the sample size is often determined to detect the target treatment effect with...
Read article
December 8, 2021
Despite the debate in the scientific community on adaptive sample size reassessment (SSR), noteworthy developments have...
Read article
December 7, 2021
Sponsors bringing a successful new product to market have to overcome two hurdles: submission and reimbursement. For...
Read article
November 30, 2021
The FDA “Real-Time Oncology Review (RTOR)”[1] is an “FDA project started in 2018 to facilitate earlier submission of...
Read article
November 30, 2021
The aim of any clinical research is to detect the actual difference in treatment effect between two groups (power) and...
Read article
November 23, 2021
A randomized clinical trial (RCT) is the gold standard approach to demonstrate the efficacy and safety of novel...
Read article
November 17, 2021
Traditionally, the teams responsible for clinical development and regulatory submissions do not consult the market...
Read article
November 15, 2021
Since its first publication ten years ago, Cyrus Mehta and Stuart Pocock’s Promising Zone Design for sample size...
Read article
November 11, 2021
The value of Real World Evidence (RWE) is well known to many stakeholders, but its full potential for market access and...
Read article
November 10, 2021
There are many reasons why traditional approaches to designing a clinical study are generally suboptimal and do not...
Read article
November 8, 2021
Ten years ago Cytel co-founder Professor Cyrus Mehta and Professor Stuart Pocock of the London School of Hygiene and...
Read article
November 4, 2021
The prominent European conference for Health Economics and Outcomes Research (HEOR), ISPOR Europe 2021, is around the...
Read article
November 2, 2021
A good clinical study design performs well not only under the ideal target scenario. Statisticians should be able to...
Read article
October 27, 2021
A staged investment strategy aligns R&D decisions and financial planning with the interim looks of a clinical trial. If...
Read article
October 22, 2021
Currently, there are many treatment options for Cancer such as, Immunotherapy, Radiation Therapy, Chemotherapy etc. If...
Read article
October 21, 2021
A number of methods currently exist to measure non-adherence and non-persistence of medical therapies, for improved...
Read article
October 20, 2021
For health innovators, trial selection is a key success factor as there are no second chances. But how do you find the...
Read article
October 14, 2021
When choosing the optimal clinical trial design for a given study, sponsors face critical questions like choice of...
Read article
October 7, 2021
‘Drugs do not work in patients who do not take them,’ said former surgeon general C. Everett Koop. Unfortunately, the...
Read article
October 6, 2021
Master Protocols are advanced and innovative clinical trial designs that can evaluate multiple therapies and disease...
Read article
October 5, 2021
Traceability is crucial in all steps of clinical data handling, from data collection to final analysis. The importance...
Read article
October 1, 2021
The COVID-19 pandemic elevated the challenge of designing and executing clinical trials within a substantially...
Read article
September 30, 2021
A complex methodological issue which arises in the production of real-world evidence involves the degree to which...
Read article
September 29, 2021
While there is a plethora of rare diseases, some 7000 diseases and counting, one needs to consider the statutory...
Read article
September 24, 2021
We may be familiar with adaptive designs, but their complexity has made them difficult to implement and their benefits...
Read article
September 23, 2021
A complex methodological issue which arises in the production of real-world evidence involves the degree to which...
Read article
September 21, 2021
Earlier this summer, we published a series of articles on the need to utilize weighting and prioritization tools in the...
Read article
September 15, 2021
There is recognized heterogeneity within any given tumor‐type from patient to patient (inter‐patient heterogeneity),...
Read article
September 14, 2021
Alyssa Biller is a Senior Solutions Consultant at Cytel and currently plays a primary role in onboarding customers for...
Read article
September 8, 2021
A key decision in the design of clinical trials in oncology involves whether to select progression free survival (PFS)...
Read article
August 31, 2021
For years, I have been telling the recruiters at Cytel to be wary of candidates claiming to have a CDISC Certification...
Read article
August 20, 2021
In clinical research, decision makers need to choose among different courses of action every day. Whether it is...
Read article
August 18, 2021
During the design of a clinical trial, many biotechs want to substantially reduce the risk of a good new therapy being...
Read article
August 13, 2021
With the cloud computing power that we have today, we can run simulation of 1000s of designs with each design...
Read article
August 9, 2021
Recently we discussed examining clinical development through a Bayesian lens, in honor of Cytel co-founder Nitin Patel...
Read article
August 5, 2021
It is important to compare competing interventions to determine value of medicines, both from clinical and societal...
Read article
July 30, 2021
I had recently (for the first time) the pleasure and honor to attend a virtual meeting with the FDA, a pre-NDA Type-B...
Read article
July 28, 2021
“We found an optimal design in hours that might have taken months to find using standard methods,” reflected Fabien...
Read article
July 27, 2021
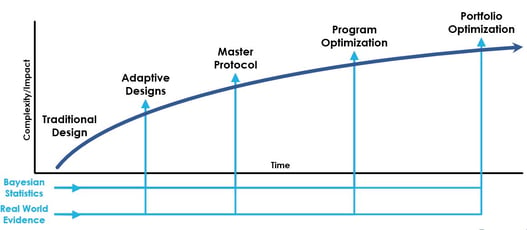
Program and portfolio optimization creates a framework throughout the course of the clinical development journey, that...
Read article
July 22, 2021
Earlier this year, Cytel founder Cyrus Mehta observed that clinical trial design is often treated like an art rather...
Read article
July 21, 2021
Conducting a transparent, targeted and robust benefit-risk assessment of a new drug or product is one of the most...
Read article
July 20, 2021
Anil Golla is Vice President, Functional Service Provision (FSP) at Cytel. After 17 years of working at pharmaceutical...
Read article
July 14, 2021
For decades, statisticians have cultivated methods to optimize and de-risk clinical trials for strong regulatory...
Read article
July 7, 2021
Suppose a statistician were to tell a clinical trial sponsor that it was possible to improve the power of the sponsor’s...
Read article
July 2, 2021
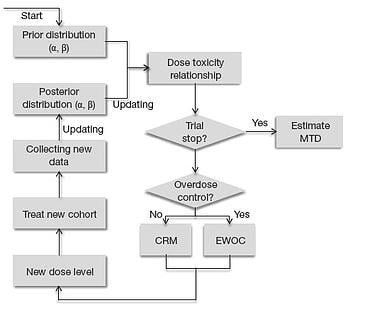
Popular statistical designs, such as CRM (O’Quigley et al., 1990), mTPI-2 (Guo et al., 2017), and i3+3 (Liu et al.,...
Read article
July 1, 2021
Synthetic control arms (SCAs) are best suited for situations when a single arm trial is run in a patient population...
Read article
June 30, 2021
When Cyrus Mehta introduced the Promising Zone Design over a decade ago, the new statistical method not only...
Read article
June 25, 2021
The main challenge associated with the development of therapies for rare diseases is typically the small study sample...
Read article
June 24, 2021
Synthetic control arms (SCAs) leverage real world data from various sources or evaluations of historical clinical data...
Read article
June 23, 2021
Much of the discussion about clinical trial design considers methods to optimize performance characteristics and...
Read article
June 18, 2021
Next week’s PSI Conference will feature Dr. Yannis Jemiai speaking on the use of Scoring Functions in the re-imagined...
Read article
June 17, 2021
In Part 2 of this four-part blog series, we bust another myth surrounding synthetic control arms (SCAs) used in...
Read article
June 15, 2021
Ten years ago, a seminal paper published by Cytel Founder Cyrus Mehta, introduced the Promising Zone Design to...
Read article
June 14, 2021
ACDM’s (Association for Clinical Data Management) eDigital data management expert group (DMEG) focuses on driving...
Read article
June 10, 2021
Over the past decade, single arm trials have emerged as an accepted way of assessing a new treatment intervention....
Read article
June 9, 2021
When selecting clinical trial designs, how many design options should a sponsor explore? Would a sponsor feel more...
Read article
June 4, 2021
Bayesian methods have been playing a key role in transforming clinical research in therapeutic areas such as oncology...
Read article
June 3, 2021
Bayesian methods allow for the incorporation of prior knowledge, in terms of either expert opinion from clinicians or...
Read article
June 2, 2021
For many decades the Pareto Frontier has been employed by actors in the private sector to evaluate and understand the...
Read article
May 28, 2021
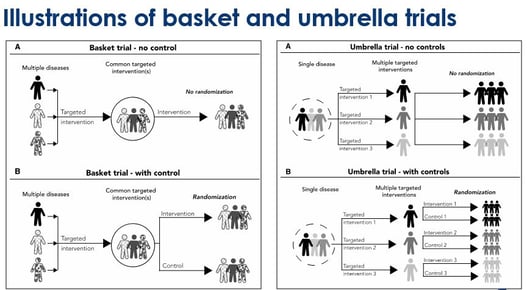
Advancements in biomarkers and momentum in precision medicine has paved the foundation for complex studies like basket...
Read article
May 20, 2021
As more payers and HTA agencies turn to real world data to compare the effectiveness of various treatment effects, two...
Read article
May 19, 2021
Clinical trial sponsors are more likely than ever to use the power of simulation and forecasting to evaluate the...
Read article
May 14, 2021
When developing clinical strategy, applying familiar business principles to the specific requirements of clinical...
Read article
May 13, 2021
Cytel’s HEOR and RWE Expertise has grown quite significantly in the past year. Could you speak a little bit about the...
Read article
May 12, 2021
Did you know that Bayesian methods can strengthen Frequentist trials through the use of Bayesian decision criteria or...
Read article
May 7, 2021
When is a study design considered to be optimal? A good design performs well not only under the ideal target scenario...
Read article
May 5, 2021
Cytel’s team of HEOR and Real-World Evidence (RWE) experts are all set to make a splash at the Virtual ISPOR on May 17...
Read article
April 29, 2021
After twenty years in pharma, Anna Forsythe was frustrated by traditional vendors who used outdated methods to prepare...
Read article
April 28, 2021
Recently, Cytel co-founder Professor Cyrus Mehta noted that, “Clinical trial design selection is too much like an art,...
Read article
April 26, 2021
As we all continue to take necessary precautions against the spread of COVID-19 virus, this year again the CDISC EU...
Read article
April 23, 2021
The urgent need to discover and assess the efficacy and safety of COVID-19 vaccine candidates will affect the future...
Read article
April 21, 2021
Two therapies are placed in head-to-head clinical trials when they are compared against each other as opposed to a...
Read article
April 20, 2021
For much of the past three decades, even as methodologies for clinical trial design have advanced and refined, the idea...
Read article
April 14, 2021
In the era of modern clinical development, several companies are turning towards novel and innovative approaches such...
Read article
April 13, 2021
There has been an increasing use of digital measures in drug development recently. New wearables technologies can help...
Read article
April 7, 2021
Scientists at Cytel recently published a paper in the Journal of the American Medical Association (JAMA). Among the...
Read article
April 2, 2021
Earlier this month, my colleagues at Cytel Canada published a paper in JCO Clinical Cancer Informatics, offering a...
Read article
March 31, 2021
As decentralized clinical trials become more attractive in an era of COVID-19, the role of wearables in clinical...
Read article
March 30, 2021
As of today, our Industry has not defined any approach, nor does an official regulatory agency...
Read article
March 26, 2021
Former Commissioner of the FDA, Dr. Scott Gottlieb, in several public presentations, would bemoan missed chances to...
Read article
March 25, 2021
Wearables have experienced increasing applicability within medical device trials, yet the regulations for the use of...
Read article
March 19, 2021
The use of wearable and digital technology requires considerations for both drugs and devices regulations, and...
Read article
March 18, 2021
Over the past ten years High-Performance Computing (HPC) has transformed medical research through advances in genomics,...
Read article
March 12, 2021
With wearables likely to become a regular part of clinical trial design, statisticians could benefit by familiarizing...
Read article
March 11, 2021
In the recent years, Oncology trials are seeing a technological shift that is expected to make them faster and more...
Read article
March 9, 2021
When statistical sciences were in their infancy, the communicative benefits of statistics were widely touted. Thousands...
Read article
March 5, 2021
Early stage Phase 2 clinical trials are often designed as multi-stage single arm trials, which quickly identify...
Read article
February 26, 2021
In the world of clinical trials, the pace of innovation is accelerating, and approaches such as Bayesian methods are...
Read article
February 25, 2021
The convergence of several distinct trends has made wearables an increasingly attractive option for use in confirmatory...
Read article
February 24, 2021
In a previous post, I discussed the importance of proper use of CDISC Controlled Terminology (CDISC CT) in SDTM....
Read article
February 19, 2021
C-Suite and R&D Decision-Makers are always striving to make evidence-driven decisions. Yet the rules by which evidence...
Read article
February 18, 2021
In the last few years, there has been a growing interest in historical borrowing or augmented trials. There is an...
Read article
February 17, 2021
The COVID-19 Pandemic prompted the rapid surge in the generation of clinical data that has been scattered across...
Read article
February 12, 2021
In recent times, Single arm trials are being increasingly used to assess new treatment interventions. They establish...
Read article
February 11, 2021
The past two years have witnessed a heightened interest in the use of wearables in clinical development. The unexpected...
Read article
February 4, 2021
The past decade has witnessed the rise of simulations-based clinical trial optimization in a manner unimaginable to...
Read article
February 2, 2021
As the use of master protocols becomes more prevalent in drug development, Bayesian methods are extensively used to...
Read article
February 1, 2021
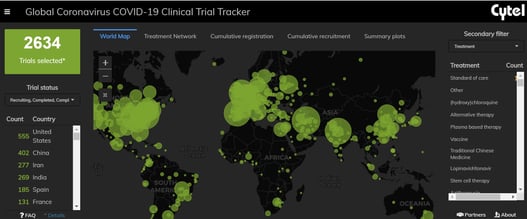
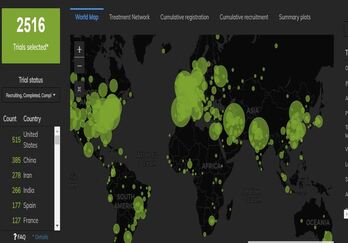
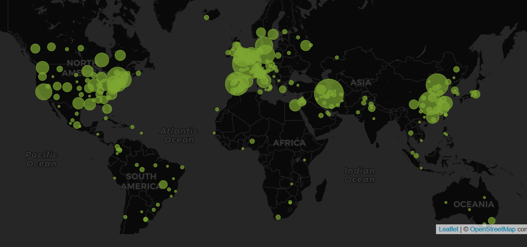
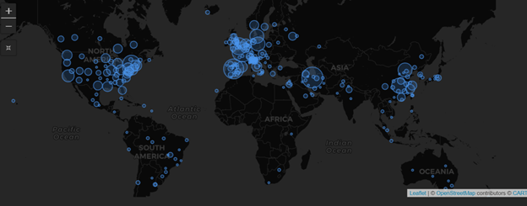
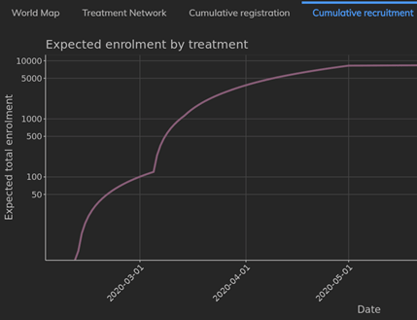
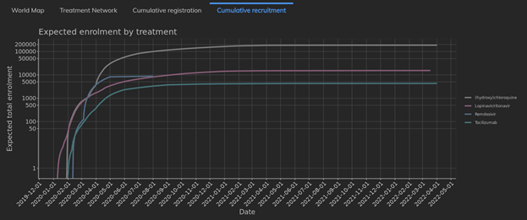
Cytel’s COVID-19 Trial Tracker continues to provide real time updates to the status of COVID-19 clinical trials...
Read article
January 29, 2021
Historically, advances in the statistical design of clinical trials have accompanied progress within the science and...
Read article
January 28, 2021
The Christmas break presented an opportunity to make my first concrete steps into the CDISC Library. Overall, it was a...
Read article
January 26, 2021
Bayesian models offer a flexible way of incorporating historical controls in the analysis of trial data (whether single...
Read article
January 22, 2021
COVID-19 has transformed the pharmaceutical industry in a manner that few could have predicted only a year ago. One of...
Read article
January 20, 2021
One of the most difficult challenges facing Research and Development teams involves determining how to make tradeoffs...
Read article
January 19, 2021
As we enter 2021 with new COVID-19 vaccines and greater optimism about the pipeline of drugs and devices positioned for...
Read article
January 13, 2021
Effective use of the right outsourcing solution can enable sponsors to respond to market needs and change course where...
Read article
January 12, 2021
The rapid pace of technology has opened up numerous avenues for advanced innovative clinical trial design, but how can...
Read article
January 11, 2021
In April 2020, Cytel launched an open-access global COVID-19 Clinical Trial Tracker to help facilitate greater...
Read article
December 22, 2020
At Cytel, we have been diligently working to become an organization deeply committed to uplifting and enriching...
Read article
December 21, 2020
2020 has been an unusually difficult year as the global pandemic impacted all of our lives. This year, the Cytel blog...
Read article
December 18, 2020
Can I submit software programs other than SAS? What software programs should I submit? Are sponsors required to submit...
Read article
December 17, 2020
As Chief Scientific Officer, Dr. Yannis Jemiai plays a pivotal role in maintaining Cytel’s well-established reputation...
Read article
December 16, 2020
As we prepare to close the door on 2020, we asked Pantelis Vlachos, Principal/Strategic Consultant for Cytel, to share...
Read article
December 15, 2020
When designing clinical trials, biostatisticians and clinical development teams are often faced with a conundrum. Given...
Read article
December 10, 2020
Sachin Sobale began his career with Cytel as a young statistician. He has been associated with the company for more...
Read article
December 9, 2020
As a part of Cytel’s Advanced Design Framework, a new Framework for the statistical design of clinical trials, Cytel...
Read article
December 8, 2020
The Cytel COVID-19 Trial Tracker brings you an up to the minute, real time dashboard about COVID-19 trials around the...
Read article
December 3, 2020
Increasing Clinical Development Productivity Using Statistics and Cloud-Computing The need for Re-imagining Clinical...
Read article
December 1, 2020
An extraordinary amount of global research is underway as the COVID-19 pandemic continues to evolve and spread. As...
Read article
November 24, 2020
Virtual ISPOR 2020, held November 16 to 19, presented new opportunities for scientific interaction amongst HEOR...
Read article
November 23, 2020
The Virtual PHUSE-EU CONNECT Conference was held from November 8 to 13 and the event was a great success, despite all...
Read article
November 19, 2020
Data Monitoring Committees (DMCs) are groups of independent experts who periodically receive (by-arm) reports created...
Read article
November 18, 2020
The current state of the clinical trials industry faces a challenge that was only hypothetical three or four years ago....
Read article
November 17, 2020
As a part of Cytel’s "New Horizons Webinar Series", Alind Gupta, Senior Data Scientist, presents case studies from his...
Read article
November 16, 2020
MUCE is a Bayesian solution for cohort expansion trials where multiple dose(s) and multiple indication(s) are tested in...
Read article
November 12, 2020
Cytel and Ingress Health (now a Cytel company) will be contributing to a range of events at Virtual ISPOR EUROPE 2020,...
Read article
November 11, 2020
The widespread use of cloud-computing has altered the clinical trial design process. Whereas three or four years ago,...
Read article
November 9, 2020
As the evolving COVID-19 pandemic continues, the Cytel COVID-19 Trial Tracker continues to bring you an up to the...
Read article
November 5, 2020
In oncology, many manufacturers go into niche indications, where there are very specific tumors, and then they opt for...
Read article
November 4, 2020
Cytel has recently revealed its Advanced Design Framework, a method developed by Cytel’s thought leaders that draws on...
Read article
November 3, 2020
When an expert statistician is paired with an experienced set of data managers, opportunities to capitalize on...
Read article
November 2, 2020
Measuring treatment effect during a clinical trial is often the source of much debate, particularly during rare disease...
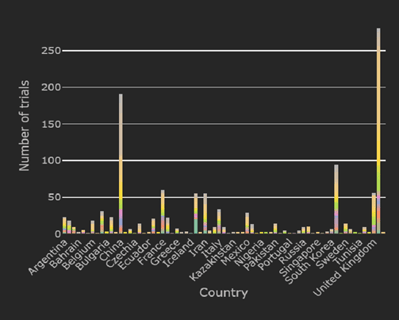
Read article
October 29, 2020
Cytel has recently revealed its Advanced Design Framework, a method developed by Cytel’s thought leaders that draws on...
Read article
October 28, 2020
About three years ago, Cytel was helping a sponsor on a project where I had to conduct surveillance of some CRO...
Read article
October 27, 2020
Cytel recently conducted a webinar on Bayesian Dose-finding Designs for Modern Drug Development, presented by Dr. Yuan...
Read article
October 26, 2020
Pharmaceutical and biotech companies are under pressure to deliver more and deliver faster with fewer resources. The...
Read article
October 23, 2020
Cytel’s New Horizons Webinar Series introduces you to the latest innovations in statistical trial design. This webinar...
Read article
October 22, 2020
In this two-part blog series, we interview Bart Heeg, Vice President HEOR and Founder at Ingress Health (A Cytel...
Read article
October 21, 2020
Cytel has recently revealed its Advanced Design Framework, a method developed by Cytel’s thought-leaders after a decade...
Read article
October 20, 2020
In this interview with Thomas Wilke, Principal Scientist at Ingress-Health (a Cytel company), we talk to him about his...
Read article
October 19, 2020
COVID-19 has created extreme uncertainties -- a dearth of historical information combined with the need for safety,...
Read article
October 15, 2020
For over a decade, advanced trial design techniques have promised efficient trials with accelerated timelines,...
Read article
October 14, 2020
A credible evidence base is needed to support and document the economic value of new technologies and therapeutic...
Read article
October 13, 2020
The combination of greater access to electronic health records, bigger electronic claims datasets, and the need for...
Read article
October 12, 2020
Cytel and Novartis are together hosting a complimentary Bayesian Virtual Symposium and an Interactive 7-part workshop....
Read article
October 7, 2020
With the rise in digital technologies, there has been an explosion in the volume and type of data sources. We can...
Read article
October 1, 2020
Methods involving Group Sequential Designs are one of the earliest deviations from a traditional two-arm clinical trial...
Read article
September 29, 2020
Innovation in trial designs are offering new routes forward for organizations of any size. They are now also aligned...
Read article
September 25, 2020
Regulators in both the United States and Europe have responded positively to the use of synthetic control arms (SCA)s...
Read article
September 22, 2020
Research Scientists, Thomas Wilke and Sabrina Mueller recently published a manuscript on “Diabetes-Related...
Read article
September 17, 2020
Keeping up with the rapid pace of clinical development means that we need to adopt the innovative or computationally...
Read article
September 16, 2020
Synthetic control arms (SCA) are virtual trial arms that use historical claims data and observational data to simulate...
Read article
September 15, 2020
In clinical trials with small or sparse data, statistical methods meant for large sample sizes may not be helpful to...
Read article
September 14, 2020
“A good start is half the battle” (the Before) when submitting data to the FDA and there are a couple of cherries to...
Read article
September 10, 2020
Single arm trials are emerging as an accepted way of assessing a new treatment intervention. They establish clinical...
Read article
September 9, 2020
Cytel’s co-founder, Nitin Patel, conducted a webinar on designing clinical trials from a program-level perspective. His...
Read article
September 4, 2020
On Friday September 11, Cyrus Mehta, co-founder of Cytel, will be delivering a talk to the Heart Failure Collaboratory,...
Read article
September 1, 2020
In this blog, Cytel's SVP Corey Dunham’s talks about our Functional Services teams and the qualities we seek in new...
Read article
August 31, 2020
Pantelis Vlachos, Principal, Strategic Consultant at Cytel, conducted a webinar to introduce the capabilities of East...
Read article
August 26, 2020
As Chief Scientific Officer, Dr. Yannis Jemiai plays a pivotal role in maintaining Cytel’s well-established reputation...
Read article
August 25, 2020
It is important to take a strategic approach to clinical development in order to minimize the potential for Phase 3...
Read article
August 21, 2020
As uses of real world data become more familiar for trial design and regulatory submission, sponsors might become more...
Read article
August 18, 2020
Mrudula Joshi joined Cytel in July 2005 as a young SAS programmer. Last month, she celebrated her 15th year work...
Read article
August 12, 2020
Cytel’s Biostatistics and Statistical Programming team provides integrated solutions, by blending the expertise of...
Read article
August 11, 2020
While there is increasing optimism about the discovery of a COVID-19 vaccine, one of the less talked about aspects of...
Read article
August 7, 2020
Recently a biotech approached Cytel for support with a Phase 2 Study in oncology. Regulators had requested a natural...
Read article
August 5, 2020
Cytel is conducting two pilot projects on head-to-head comparisons using real world data. These projects in oncology...
Read article
August 4, 2020
Last year, Paul Terrill, Associate Principal of Strategic Consulting at Cytel, presented an engaging webinar on the...
Read article
July 30, 2020
A new peer-reviewed article co-authored by several Cytel scientists re-examines the way in which adaptive trials are...
Read article
July 28, 2020
CDISC standards have been around for a while with the first SDTM Standard version released in 2004. However, it was...
Read article
July 27, 2020
Cytel is conducting a webinar series on complex innovative trial designs. Dr. Thomas Burnett, Senior Research Associate...
Read article
July 23, 2020
Just as there are numerous adaptations that fall within the umbrella of adaptive designs, there are several different...
Read article
July 22, 2020
Cytel brings to you a new blog series on technology and Bayesian decision-making by Pantelis Vlachos,...
Read article
July 15, 2020
Unlike many therapeutic areas, oncology benefits from having standardized endpoints like overall survival and...
Read article
July 14, 2020
Cytel SVP Corey Dunham’s inaugural post on leading the industry’s largest Biometrics CRO considers the FSP Engagement...
Read article
July 13, 2020
Cytel is hosting a complimentary webinar series that introduces biostatisticians and other members of the development...
Read article
July 11, 2020
Last week we challenged you to take a COVID-19 Clinical Development Quiz. Many of you used our Trial Tracker to find...
Read article
July 9, 2020
There has been an increased use of synthetic control arms for regulatory submissions in recent years, with three rare...
Read article
July 7, 2020
Having its roots in the seminar rooms of the Dana Farber Cancer Institute, Cytel has a long record of establishing new...
Read article
July 2, 2020
There are currently 1570 registered clinical trials for COVID-19 therapies and vaccines. Approximately 20% are...
Read article
June 29, 2020
From the time the COVID-19 outbreak was declared a pandemic, the number of studies conducted around the world to either...
Read article
June 27, 2020
Expert statisticians at Cytel have spent the past three and a half months designing and deploying dozens of trials for...
Read article
June 22, 2020
Cytel scientists recently published a new eBook on synthetic control arms and a new scientific primer for the more...
Read article
June 18, 2020
One of the revelations of the COVID-19 pandemic is that the flexibility and potential of Bayesian designs goes far...
Read article
June 18, 2020
A number of trials recently disrupted by the COVID-19 pandemic are now in the process of re-assessing recruitment...
Read article
June 13, 2020
The Cytel COVID-19 Trial Tracker continues to bring you an up to the minute, real time dashboard about COVID-19 trials...
Read article
June 10, 2020
Cytel is conducting a webinar series that introduces biostatisticians to some of the more commonly used complex...
Read article
June 9, 2020
Trevor Mundel leads the Bill & Melinda Gates Foundation’s efforts to develop high-impact interventions against the...
Read article
June 8, 2020
Cytel has recently published a new ebook on synthetic control arms, and a new scientific primer as well.
Read article
June 4, 2020
This weekly snapshot gives you a quick briefing on the state of COVID-19 therapy and vaccines development. As we head...
Read article
May 29, 2020
At the close of May 2020, we have about 500 new trials globally but trends in trial design and choice of therapies...
Read article
May 27, 2020
In this blog, we speak with Christopher Jennison, Professor of Statistics at the University of Bath, UK. Professor...
Read article
May 22, 2020
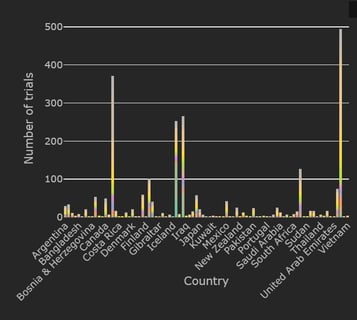
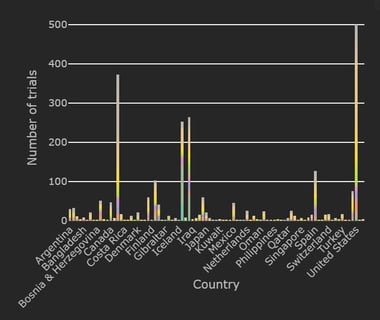
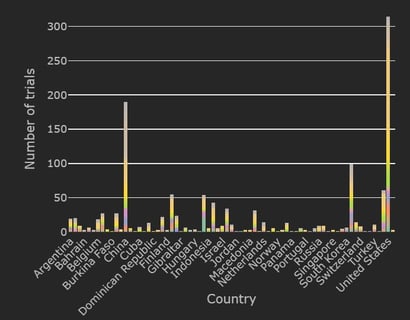
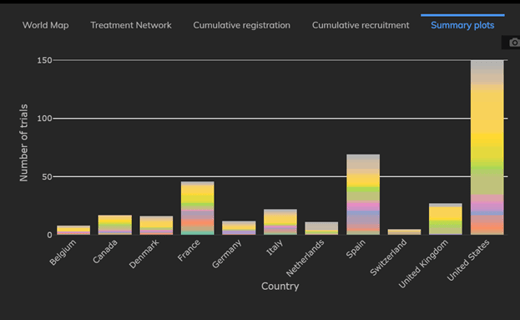
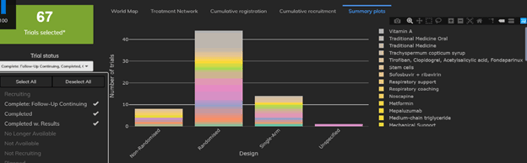
The Cytel Trial Tracker now features summary plots that display trials by country, trial status and study design. This...
Read article
May 14, 2020
This has been an exciting week for COVID-19 studies. We learned that several Cytel clients who have designed new...
Read article
May 12, 2020
In our previous blog, “Remote Working Arrangement – How to get it right?”, we talked about how the need for social...
Read article
May 7, 2020
There are now over 950 trials registered, which means that 250 new trials were registered in the past week. Only 540 of...
Read article
May 5, 2020
Cytel's team of oncology trial design and advanced analytics experts are hosting a series of complimentary webinars...
Read article
April 29, 2020
In our previous blog, we spoke with Alind Gupta, who works as a Machine Learning Researcher at Cytel in Canada. The...
Read article
April 27, 2020
In the first part of this two-parts blog, I speak about how the European CDISC Committee (E3C) together with CDISC...
Read article
April 24, 2020
In early March, when countries around the world started implementing lockdowns, the European CDISC Committee (E3C)...
Read article
April 23, 2020
Cytel is hosting a webinar, “A Clinician’s Perspective on Cancer Drugs Development”, on April 28, 2020. Our speaker,...
Read article
April 23, 2020
Every Week Cytel Brings You Further Insights from the COVID-19 Trial Tracker From April 8 through April 17, the number...
Read article
April 20, 2020
Cytel is hosting a webinar on Transparent Machine Learning in Oncology, on April 21, 2020. Our speaker, Alind Gupta,...
Read article
April 7, 2020
Cytel has industry-leading experts in Statistical Programming with years of SAS® Programming expertise and in-depth...
Read article
April 2, 2020
On March 16, the World Health Organization (WHO) Director-General, Dr. Tedros Adhanom Ghebreyesus, in his media...
Read article
March 31, 2020
Since 1953, when the discovery of the structure of DNA was made, we have seen great advancements in genomics....
Read article
March 26, 2020
Further regulatory guidance has been released concerning the implications of the Coronavirus disease (COVID-19) on...
Read article
March 23, 2020
On March 20th the European Commission, the European Medicines Agency (EMA) and the Heads of Medicines Agency (HMA)...
Read article
March 19, 2020
The FDA issued a guidance yesterday on how the COVID-19 Pandemic may affect the conduct of clinical trials. Below are...
Read article
March 5, 2020
Generating high-quality clinical data is a vital but challenging task in modern drug development. Unfortunately, in the...
Read article
February 25, 2020
Over the past decade, a new trend began to emerge, changing the way that clinical trials are conducted. Whereas...
Read article
February 25, 2020
Regulators in both the United States and Europe have responded positively to the use of SCAs in clinical...
Read article
February 20, 2020
It is widely acknowledged among drug developers that one of their most important assets is the data generated during...
Read article
January 30, 2020
The Cytel Trial Design Innovations (CTDI) Webinar Series recently hosted a webinar on designing event-based studies....
Read article
January 22, 2020
In the quest for clinical success, we all strive for evidence packages of the highest quality. If the clinical data is...
Read article
January 16, 2020
January’s Cytel Trial Design Innovations (CTDI) Webinar Series will feature Biostatistician and pioneering Bayesian...
Read article
January 8, 2020
In clinical development, data is the vital ‘foundation’ that supports your programs. To successfully bring a promising...
Read article
December 16, 2019
Cytel Inc. and Axio Research joined forces in June 2019, expanding our ability to solve the most complex analytical...
Read article
December 10, 2019
In association with Statisticians in the Pharmaceutical Industry (PSI) , UCB and Cytel hosted a symposium on September...
Read article
December 5, 2019
Author: Michael S. Paas, Market Access & Commercialization Expert, Executive at AbbVie and Guest Author at Cytel In...
Read article
December 2, 2019
Author: Michael S. Paas, Market Access & Commercialization Expert, Executive at AbbVie and Guest Author at Cytel...
Read article
November 27, 2019
At the 2019 Challenges in Rare Diseases Clinical Trials Symposium and East training, Cytel partnered with Alexion to...
Read article
November 21, 2019
Author: Michael S. Paas, Market Access & Commercialization Expert, Executive at AbbVie and Guest Author at Cytel In my...
Read article
November 14, 2019
Market access strategy is an integral part of the clinical development process to ensure success in global healthcare...
Read article
November 5, 2019
Cytel is delighted to have Kannan Natarajan speaking at the “Complex Innovative Trial Design Symposium and East User...
Read article
October 23, 2019
October 3, 2019 was an important day for the ADaM team as it marked the release of the ADaM Implementation Guidance...
Read article
October 10, 2019
A disease is generally considered to be rare if it affects one patient per 200,000 people (1) and most rare diseases...
Read article
September 12, 2019
"If you went to bed last night as an industrial company, you're going to wake up today as a software and analytics...
Read article
August 23, 2019
In place of collecting data from patients recruited for a trial who have been assigned to the control or...
Read article
August 14, 2019
This article was originally published as part of a series by pharmaphorum in association with Cytel and is reproduced...
Read article
August 7, 2019
In this blog, from our career perspectives series, we talk with Jayshree Garade Associate Director, Statistical...
Read article
August 1, 2019
The term biomarker signature describes the behavior of a set of biomarkers that define a signature to maximize the...
Read article
July 22, 2019
Health economics and adaptive design methods share common ground in that they both aim to support more efficient and...
Read article
July 8, 2019
Cytel recently hosted a very well-attended and engaging webinar on the topic of “Estimands, not just a statistical...
Read article
June 27, 2019
By Nicolas Rouillé and Eric Henniger The right design and the right data ultimately leads to the right decisions, so...
Read article
June 14, 2019
This article was originally published as part of a series by pharmaphorum in association with Cytel and is reproduced...
Read article
May 7, 2019
Nowadays, it’s difficult to pick up a mainstream newspaper or read an industry publication without seeing reference to...
Read article
April 24, 2019
In this blog, Alla Muchnik, Senior Clinical Data Manager at Cytel, discusses how specialist CROs can add value and...
Read article
April 10, 2019
In this blog, Jonathan Pritchard, Director Business Development at Cytel, draws on his experience in commercial,...
Read article
March 26, 2019
At the Partnerships in Clinical Trials Conference in Barcelona in November 2018, Strategic Consultant Ursula Garczarek...
Read article
March 20, 2019
No one plans to have a trial whose data collection needs rescuing. However, lagging enrollment rates, operational...
Read article
February 28, 2019
In honor of Rare Disease Day 2019 we share a new Cytel podcast featuring Cytel Strategic Consultant Ursula Garczarek...
Read article
February 21, 2019
In 2018, Cytel ran a qualitative survey among biostatisticians and programmers on trends in data science and...
Read article
February 8, 2019
A 2018 publication in the Biometrical Journal by Cytel’s Cyrus Mehta, Lingyun Liu and Sam Hsiao, ‘Optimal Promising...
Read article
January 10, 2019
Nand Kishore Rawat is a Director and Head, Early Phase Biostatistics based in the King of Prussia, PA Cytel office. We...
Read article
December 5, 2018
In this blog, Paul Terrill, Director of Strategic Consulting at Cytel outlines his blueprint for ensuring smooth...
Read article
November 27, 2018
This is the third in our blog series ' The Good Data Submission Doctor' in which Angelo Tinazzi, Director of Standards,...
Read article
November 12, 2018
In this blog, we talk with Robert Greene, Founder and President of the HungerNDThirst Foundation, about his upcoming...
Read article
November 8, 2018
Data is the most crucial asset in any clinical trial and is used to ultimately drive the decision-making process...
Read article
November 5, 2018
Cytel biostatisticians Cyrus Mehta and Lingyun Liu, together with Charles Theuer, CEO of TRACON Pharmaceuticals have...
Read article
November 1, 2018
In this second post of the “Good Data Submission Doctor” ( read my first post The Master Recipe: Quality and Attention...
Read article
October 23, 2018
Cytel data scientists apply advanced statistical techniques including predictive modeling of biological processes and...
Read article
October 5, 2018
We are delighted that Stephen Senn will be joining us at the EUGM on November 14th and 15th in Darmstadt, Germany. In...
Read article
October 1, 2018
One of my wife’s favorite TV shows is ‘Quattro Ristoranti’ (Four Restaurants). In each episode of the show, 4...
Read article
September 27, 2018
In this blog, we talk with Heiko Götte, Senior Expert Biostatistician at Merck about his upcoming presentation at...
Read article
September 20, 2018
At Cytel we believe that expert statistical input has the power to shape the future of clinical development: de-risking...
Read article
September 17, 2018
Immunotherapy has brought us many promises, most notably, of a future where humans are able to harness their body’s own...
Read article
September 10, 2018
The biopharmaceutical and healthcare industries now collect more data than ever before due to advances in the variety...
Read article
September 7, 2018
On August 29th 2018, the FDA announced (1) that it would be establishing a Complex Innovative Trial Design (CID) Pilot...
Read article
September 5, 2018
EnForeSys is Cytel’s tool for patient recruitment planning. We have discussed on the blog recently with Tufts...
Read article
August 31, 2018
JSM 2018, ASA’s annual gathering of over 6500 attendees attracted statisticians and data scientists to the beautiful...
Read article
August 23, 2018
Cytel has grown significantly over the last 30 years, with operations across North America, Europe, and India. All of...
Read article
August 21, 2018
After having spent 15 years in the pharmaceutical industry, in May of 2018, I decided to explore new horizons and took...
Read article
August 15, 2018
Cytel’s 7th East User Group Meeting (EUGM) will take place on November 14 & 15, 2018 at Merck in Darmstadt, Germany,...
Read article
August 1, 2018
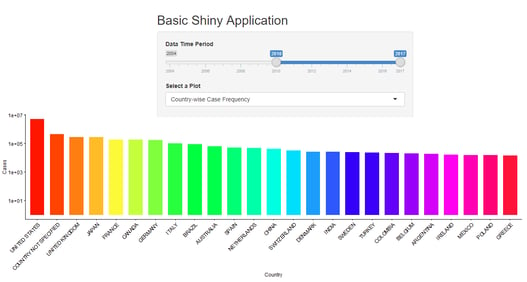
By Gordhan Bagri and Munshi Imran Hossain with H A S Shri Kishore Shiny (from RStudio) is one of the most popular R...
Read article
July 27, 2018
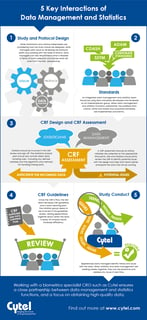
In this blog, we share a new infographic based on this popular blog post illustrating some of the critical interactions...
Read article
July 24, 2018
At Cytel our strategic consulting team works on a wide range of projects including: Identifying the best clinical trial...
Read article
July 18, 2018
A recent article published by Cytel authors Samadhan Ghubade, Sharayu Paranjpe, Kushagra Gupta, Anil Gore and colleague...
Read article
July 16, 2018
In this blog we share a case study of how we established and ramped up a functional service outsourcing partnership for...
Read article
July 3, 2018
It’s shaping up to be a busy year for Cytel’s software development team with a number of upgrades and planned launches...
Read article
July 2, 2018
A number of the Cytel team were in Amsterdam, 3rd- 6th June 2018 for the PSI Conference. This year’s conference was...
Read article
June 28, 2018
By Esha Senchaudhuri With thanks also to Jitendarreddy Seelam and Ramanatha Saralaya for their input. The fact of the...
Read article
June 19, 2018
At the recent PCMG conference in Malta, Adrian Otte ( Independent Consultant, formerly VP Global Development Operations...
Read article
June 14, 2018
In this blog, Paul Fardy, Executive Director of Data Management at Cytel shares his thoughts on how the data manager...
Read article
May 23, 2018
Our Industry Voices series showcases our clients’ innovative work and breakthrough therapeutics in oncologic...
Read article
May 16, 2018
Our Industry Voices series showcases our clients’ innovative work and breakthrough therapeutics in oncologic...
Read article
May 9, 2018
As we prepare to head to ASCO in under a month's time, we are pleased to share a new ebook that showcases some key...
Read article
April 25, 2018
Data management is an essential building block for successful Immuno-Oncology (I-O) trials. At the Immuno-Oncology...
Read article
April 18, 2018
Our recent Clinical Biometrics Survey explored the views of respondents from across the statistical programming,...
Read article
April 13, 2018
We return to our discussion with Ken Getz of the Tufts CSDD for part 2 of our blog post on key challenges in clinical...
Read article
April 5, 2018
Photo by J. Kelly Brito on Unsplash Research on clinical trial enrollment makes for sobering reading, characterized by...
Read article
April 3, 2018
By Esha Senchaudhuri In response to its R&D productivity from 2005 – 2010, AstraZeneca took the initiative in 2011 to...
Read article
March 16, 2018
At Cytel we believe that expert statistical input has the power to shape the future of clinical development: de-risking...
Read article
March 14, 2018
Photo by Agê Barros on Unsplash by Natalie Fforde, Senior Director of FSP Services at Cytel With effective use of...
Read article
March 8, 2018
Photo by Steinar Engeland on Unsplash By Ivan Navarro, Data Scientist at Cytel R is an open-source implementation of...
Read article
March 6, 2018
Photo by Dose Media on Unsplash By Charles Liu, Senior Product Manager, Cytel Several years ago, I was one of few...
Read article
February 28, 2018
There is a consensus in the industry that data on rare diseases is limited, incomplete, and difficult to find or...
Read article
February 27, 2018
We extend our congratulations to Lipopharma and the CLINGLIO project consortium on their recent 6,15M€ grant award by...
Read article
February 21, 2018
By Diganta Bose, Statistical Programming Team Lead at Cytel Editor's note: This blog is based on work presented at...
Read article
February 15, 2018
A recent publication in Biometrics ‘A Gatekeeping Procedure to Test a Primary and a Secondary Endpoint in a Group...
Read article
February 13, 2018
Our strategic consulting team work on projects such as: Identifying the best clinical trial design, implementing...
Read article
February 6, 2018
We were excited to learn recently that Ajay Sathe, the CEO of our India Operations, was awarded lifetime honorary...
Read article
January 23, 2018
News Medical interviewed Dr. Rajat Mukherjee, Statistician, and Director of Data Science at Cytel to investigate the...
Read article
January 15, 2018
The problem of feature selection The explosion in the availability of big data has made complex prediction models a...
Read article
January 9, 2018
Our Career Perspectives' series is back! Cytel has industry-leading experts in statistical programming with years of...
Read article
December 12, 2017
Signal management is one of the most audited pharmacovigilance processes. It also generates one of the highest findings...
Read article
November 28, 2017
In this blog we turn to some reading matter, and interview Gautier Paux and Alex Dmitrienko about the recent book...
Read article
November 22, 2017
Cytel offers a full range of clinical data management services and the team of experts is spread across the globe. In...
Read article
November 15, 2017
Each year Halloran Consulting Group hosts‘CORE’ (Clinical Operations Retreat for Executives) as a forum for industry...
Read article
November 9, 2017
In this blog we are excited to unveil a new project which we have been hard at work on over the last few months. 2017...
Read article
October 31, 2017
As part of Cytel's new Trial Innovations Webinar Series, Pat Mitchell, Statistical Science Director at AstraZeneca...
Read article
October 27, 2017
In this blog, we share the second part of our interview with Bob Beckman, about a design concept for a confirmatory...
Read article
October 18, 2017
Cytel's new Trial Innovations Webinar Series provides a platform for the most promising new statistical approaches...
Read article
September 29, 2017
Namrata Deshpande, Senior Team Lead will be participating in a round table discussion at the Women in Statistics event...
Read article
September 20, 2017
In this blog we share a case study of work our strategic consulting team conducted supporting accurate event prediction...
Read article
September 18, 2017
By Ashwini Joshi For small sample data or rare events data, exact non-parametric tests perform better than asymptotic...
Read article
September 13, 2017
Antibiotic resistance is one of the greatest challenges facing human health today. We are excited to welcome Dr. Scott...
Read article
September 11, 2017
At the East User Group meeting (EUGM) on 25th and 26th October, we will welcome a number of renowned industry speakers...
Read article
September 7, 2017
By Munshi Imran Hossain, Software Affiliate at Cytel Biomedical signals are electrical signals collected from the body....
Read article
August 2, 2017
As a biometrics -focused CRO, Cytel regularly works across a program of studies, providing data consistency, and...
Read article
July 26, 2017
Sadly, clinical development of anti-cancer therapeutics faces particularly high rates of failure, even in the context...
Read article
July 19, 2017
With adaptive and innovative trial designs on the rise, operational implementation of interim analyses, including...
Read article
July 13, 2017
By Tejasweeni Rajput It’s been known for centuries that a picture can tell a thousand words. In an era of new...
Read article
July 11, 2017
The Adaptive Designs and Multiple Testing Procedures Workshop (ADMTP), the first joint meeting of the Adaptive Designs...
Read article
July 6, 2017
By Esha Senchaudhuri The ethical benefits of adaptive clinical trials have been widely acclaimed: higher prospects for...
Read article
June 28, 2017
About the Author: Manjusha Gode has over 28 years' IT experience spanning delivery Management, quality management,...
Read article
June 12, 2017
The Population Approach Group in Europe (PAGE) represents a community with a shared interest in data analysis using the...
Read article
June 8, 2017
Cytel's Madhusmita Panda presented at this year’s PSI Conference in the Innovative Methodology session on the topic of...
Read article
May 30, 2017
A recent paper The case for Bayesian methods in benefit-risk assessment: Overview and future directions (1) co-authored...
Read article
May 26, 2017
PharmaSUG 2017 proved to be an inspirational and informative event. With over 200 paper presentations, posters, and...
Read article
May 22, 2017
James (Jim) Bolognese, Senior Director, Strategic Consulting, Clinical Services at Cytel Inc. was named a 2017 fellow...
Read article
May 17, 2017
This new case study shares how Cytel supported a specialist biopharmaceutical company from Phase 2 trial design through...
Read article
May 4, 2017
As a recognized expert in adaptive trials, Cytel has extensive experience designing and managing trials with interim...
Read article
April 28, 2017
In this blog we share a case study in which our statistical consulting team helped a client redesign an oncology...
Read article
April 25, 2017
At a recent conference Adam Hamm, Director Biostatistics at Cytel, presented his thoughts on Best Practices and...
Read article
April 11, 2017
In a January 2017 paper (1), the FDA reviewed 22 case studies where promising Phase 2 trials did not result in...
Read article
April 5, 2017
Statistical programmers at all levels can make a significant impact on streamlining delivery, improving efficiency, and...
Read article
March 29, 2017
With an increasing interest in platform designs and other innovative designs that involve multiple comparisons over...
Read article
March 24, 2017
Robust go/no-go (GNG) decision-making is essential for effectively managing risk across a clinical portfolio. In early...
Read article
March 20, 2017
In the randomized clinical trial (RCT), the process of deciding the randomization method and implementing is critically...
Read article
March 15, 2017
A precise and thorough approach to planning is key for success in data management. The Data Management Plan (DMP) is a...
Read article
March 10, 2017
At the recent Biosimilars Summit in Philadelphia, Cytel's Pantelis Vlachos presented on statistical challenges and...
Read article
March 2, 2017
We continue our case study series with this example of a Phase 3 design that uses Bayesian decision making combined...
Read article
February 27, 2017
It’s been hard to miss the prevalence of estimand-related discussions in the last year. This is a topic which is very...
Read article
February 21, 2017
In a previous blog, we provided an overview of basic data structures in R. In this follow up piece, we will provide a...
Read article
February 15, 2017
Outsourcing solutions should never be a one size fits all process, and smaller and emerging biopharma companies may...
Read article
February 9, 2017
In this blog, Adam Hamm, PhD, Director Biostatistics at Cytel shares some of the most important knowledge he uses in...
Read article
February 6, 2017
CDISC is a global, nonprofit charitable organization whose mission is ‘to inform patient care and safety through higher...
Read article
January 30, 2017
Single ascending dose (SAD) and multiple ascending dose (MAD) studies are typically the first in human studies. They...
Read article
January 23, 2017
As a group, Cytel had over 40 successful regulatory interactions last year, many of which supported approvals for...
Read article
January 19, 2017
R is on the rise in biopharma, and as we have previously discussed on the blog, it is now time for SAS programmers to...
Read article
January 16, 2017
The Global Cardiovascular Clinical Trialists Forum is a key event bringing together leading experts from across the...
Read article
January 5, 2017
Nonlinear Mixed Effects Modeling (NONMEM) is a type of population pharmacokinetics/pharmacodynamics (popPK/PD) analysis...
Read article
December 21, 2016
December 18th 2016 was a significant date for the pharmaceutical industry and regulatory submissions. For trials which...
Read article
December 20, 2016
Data Standards play a crucial role in structuring and promoting long term value of clinical data. Clinical Data...
Read article
December 15, 2016
In April, we interviewed NIHR research fellow Munya Dimairo about the paper, ‘Adaptive designs undertaken in clinical...
Read article
December 13, 2016
In the complex world of trial design and data analysis biostatisticians and data scientists need to ensure they are...
Read article
December 6, 2016
The management of quality clinical data collection is built on a number of core essentials- including project...
Read article
December 2, 2016
At a recent Pfizer/ Cytel seminar on rare disease and oncology development, Cytel’s Lingyun Liu presented innovative...
Read article
November 21, 2016
While adaptive designs can deliver significant benefits to clinical development- including ethical benefits for...
Read article
November 18, 2016
Challenge Our client had the following key questions which they wanted our pharmacometrics group to address for an...
Read article
November 14, 2016
In our last blog, we shared some of Angelo Tinazzi and Cedric Marchand's recommendations on how to ensure independence...
Read article
November 11, 2016
A solid and robust QC process is one vital component of ensuring quality programming delivery. Angelo Tinazzi and...
Read article
November 9, 2016
Adaptive sample size re-estimation designs are an important part of the statistician's toolkit. In this first in a...
Read article
November 1, 2016
Unlike statistics which has been around in some form for hundreds of years, pharmacometrics is, by comparison, a...
Read article
October 25, 2016
Use of R is a hot topic among statisticians and programmers in the pharmaceutical industry. At the recent PhUSE...
Read article
October 13, 2016
Statistical programmers play a key role in turning the data from clinical trials into knowledge and supporting the...
Read article
October 11, 2016
Its important to take a strategic approach to clinical development in order to minimize the potential for Phase 3...
Read article
October 7, 2016
Adaptive designs have the potential to accelerate clinical development, and improve the probability of trial success....
Read article
October 5, 2016
While some progress has been made in terms of scientific development in Neuroscience and Neuropsychiatry indications,...
Read article
September 29, 2016
A paper "Best practices case studies for 'less well-understood' Adaptive designs", has been published by the DIA...
Read article
September 27, 2016
The Lung-MAP trial is an innovative biomarker driven 'precision medicine' study which evaluates five novel agents for...
Read article
September 20, 2016
Drug development is an expensive and risky business. To maximize a compound’s ultimate chances of commercial as well as...
Read article
September 13, 2016
Exposure-response data gained from clinical studies can provide a basis for model-based analysis and simulation,...
Read article
September 9, 2016
Challenge: Our client, an emerging biotechnology company, was preparing for the next stage of development for their...
Read article
September 7, 2016
In the 2010 draft FDA ‘Guidance for Industry on Adaptive Design Clinical Trials for Drugs and Biologics', the agency...
Read article
August 31, 2016
Traditional rule-based approaches to dose escalation such as 3+3 are widely used in early clinical development. They...
Read article
August 26, 2016
At the recent JSM in Chicago, Cytel’s Sam Hsaio and Lingyun Liu alongside Genentech's Romeo Maciuca, presented a...
Read article
August 23, 2016
At the recent JSM meeting in Chicago, Cytel's Jim Bolognese presented the results of work he has conducted evaluating...
Read article
August 17, 2016
Following the recent publication of their review article Adaptive Designs for Clinical Trials in the New England...
Read article
August 9, 2016
At the recent JSM Meeting, Cytel’s Yannis Jemiai presented the case study of the VALOR trial which used a promising...
Read article
August 2, 2016
Editor's note( this blog was refreshed in April 2018) As CDISC compliant submissions become increasingly expected,...
Read article
July 28, 2016
To close a clinical database right the first time you have to begin with study start-up. Clearly, you can’t close a...
Read article
July 25, 2016
Statistical programmers are in high demand within the biopharmaceutical industry, and within the dynamic world of...
Read article
July 21, 2016
Did you miss our webinar on Single and Dual Agent Dose escalation designs earlier in the year? In this blog we have...
Read article
July 18, 2016
In order for adaptive designs to reach their potential, it’s critical that knowledge is effectively dissemirnated...
Read article
July 12, 2016
How do you go about selecting the best Electronic Data Capture (EDC) system for your study? There is now a vast amount...
Read article
July 7, 2016
Why do we do what we do? At Cytel we have always been driven to deliver benefits in the service of human health, and...
Read article
July 5, 2016
At a recent PhUSE SDE, Cytel’s Chitra Tirodkar presented how East PROC MCPMod could be used to help solve the problem...
Read article
June 23, 2016
Vendor selection is a critical component of ensuring clinical trial success. A 2015 report (1) suggested that clinical...
Read article
June 21, 2016
Sunil is an Associate Director of Statistical Programming at Cytel and has over twenty-five years of experience in the...
Read article
June 16, 2016
Predicting the course of a clinical trial is something which people will always want to do-whether for statistical...
Read article
June 14, 2016
At Cytel, we are very often asked to get involved in DMCs ( Data Monitoring Committees) in a variety of capacities. Our...
Read article
June 7, 2016
At the recent CMO Summit East James ( Jim) Bolognese, Cytel’s Senior Director of Strategic Consulting, and Lou...
Read article
May 31, 2016
Last week, we were delighted to announce the release of East 6.4 bringing further cutting –edge approaches to the East...
Read article
May 19, 2016
The explosion in healthcare information and “big data “has been one of the most written about topics in the last few...
Read article
May 17, 2016
Cytel participated at PharmaSUG 2016 in Denver recently. A key event on the statistical programming global calendar,...
Read article
May 12, 2016
Once upon a time Hansel and Gretel laid a trail of breadcrumbs which they followed to find their way back home. Their...
Read article
May 10, 2016
We were fortunate to welcome Björn Bornkamp of Novartis to the EUGM 2016 presenting work he has developed jointly with...
Read article
April 28, 2016
Adaptive Designs in Practice: Interview with NIHR Research Fellow Munya Dimairo NIHR and University of Sheffield...
Read article
April 26, 2016
In this blog we’ll highlight some unique challenges that are encountered from a Data Management perspective when...
Read article
April 22, 2016
FDA draft guidance on “Co development of two or more unmarketed investigational drugs for use in combination” notes...
Read article
April 20, 2016
During the course of any clinical trial, there are often data which, while collected electronically, are outside of the...
Read article
April 15, 2016
It's critical for biostatistics and data management to be closely aligned and working effectively together. The...
Read article
April 13, 2016
We continue our series of blogs covering the expert presentations from the EAST User Group Meeting. Consultant Claire...
Read article
April 7, 2016
Francois Beckers, Global Head of Biostatistics & Epidemiology at Merck KGaA joined us at the East User Group Meeting in...
Read article
April 5, 2016
Cost of pharmaceutical development and R&D productivity is an ongoing industry concern, consistently discussed in the...
Read article
March 29, 2016
On March 16th and 17th the 5th East User Group Meeting took place in London. This very successful 2 days saw a variety...
Read article
March 23, 2016
“How many statisticians does it take to ensure at least a 50% chance of a disagreement about p-values?” So questions...
Read article
March 11, 2016
There has been increasing interest in multi-arm multi-stage trials with treatment selection and sample size...
Read article
March 8, 2016
Data standardization is critical to ensure successful regulatory submissions. While many sponsors now choose to create...
Read article
March 3, 2016
'The aim of a discussion should not be victory but progress.' This principle, expressed by the French essayist Joseph...
Read article
February 26, 2016
Remember the early days of Electronic Data Capture? Those first systems, which were revolutionary for their time...
Read article
December 3, 2015
On Dec. 12, 2015 Sangeetha Gandepalli, a Technical Lead in Clinical Data Management at Cytel, will speak before SCDM...
Read article
December 1, 2015
Risk based monitoring is a strategic monitoring practice which aims to shine the spotlight on problematic study areas,...
Read article
November 12, 2015
Last week Cytel joined forces with Sanofi/Genzyme to devote a full day of workshops and talks related to modern methods...
Read article
November 10, 2015
Last week Cytel joined forces with Sanofi/Genzyme to devote a full day of workshops and talks related to modern methods...
Read article
November 5, 2015
Oftentimes people perceive a tradeoff between speed and quality. The faster you do something the more likely you are to...
Read article
November 3, 2015
QPP (sometimes called QP2) remains at the heart of model based drug development. Short for Quantitative Pharmacology &...
Read article
October 29, 2015
A common challenge of working with small sample sizes is determining proper bias correction methods when evaluating a...
Read article
October 27, 2015
Here at Cytel we have enjoyed following the debates on the p-value controversy currently taking place on the ASA...
Read article
October 19, 2015
It is often necessary to pool safety data from late phase studies, in preparation for regulatory submission. Some of...
Read article
October 15, 2015
The method for dose-response modeling that is widely called MCPMod allows a sponsor to measure the likelihood that...
Read article
October 13, 2015
In honor of World Obesity Day (celebrated on Oct. 11 2015) here is an American Heart Association Statistical Primer on...
Read article
October 8, 2015
A recent American Statistical Association conference featured a town hall meeting to discuss the role of the...
Read article
October 5, 2015
One consideration every sponsor of a biomarker-stratified confirmatory trial must take into account, is whether to...
Read article
October 1, 2015
This week marks the sixth annual American Conference on Pharmacometrics, held this year in Crystal City, VA. Situated...
Read article
September 29, 2015
The Head of the DIA’s Adaptive Design Working Group Asks Us to Consider 5 ‘Soft-Skills’ All Effective Statisticians...
Read article
September 24, 2015
Earlier this week, Patti Arsenault, Cytel’s Global Head of Clinical Data Management, sat on an SCDM panel with members...
Read article
September 17, 2015
Most people familiar with adaptive clinical trial designs are familiar with those statistical designs that reject the...
Read article
September 4, 2015
Our Client's Challenge: Can knowledge of the relationship between biomarkers and clinical endpoints help us to optimize...
Read article
September 1, 2015
If you’re in the practice of conducting early phase clinical trials, you’ve probably heard that modern trial designs...
Read article
August 13, 2015
Full service or specialized? Full service or specialized? For many looking to hire a CRO, the answer is obvious....
Read article
August 10, 2015
When approaching a Phase 3 clinical trial, the need to ‘de-risk’ the massive investment often leads sponsors on a quest...
Read article
July 27, 2015
We were saddened to learn earlier this year, of the passing of Professor David Sackett. Widely recognized as the father...
Read article
July 21, 2015
MCP-Mod methodology for dose-ranging clinical trials has been gaining popularity since the 2013 publication of the...
Read article
July 14, 2015
When conducting Maximum Likelihood Estimation, it is assumed that the maximum likelihood estimate follows a normal...
Read article
June 26, 2015
Powering a trial for superiority can be financially risky. In some instances it may also prove unnecessary.
Read article
June 25, 2015
When conducting a clinical trial with small or sparse data sets, statistical methods meant for large sample sizes may...
Read article
June 18, 2015
In August 2006 AstraZeneca completed the ARISE trial, which aimed to determine whether AGI-1067 was effective in...
Read article
June 11, 2015
When the FDA first began to require pharmaceuticals to perform cardiovascular outcome trials to establish the safety of...
Read article
June 5, 2015
Not long ago, one of our clients submitted Phase 2 and Phase 3 data for a new rare disease drug which had received...
Read article
June 4, 2015
Two insightful papers from Applied Clinical Trials should be of interest to many clinical trial planners. The first by...
Read article
May 28, 2015
Risks in drug development range from taking the wrong drugs forward to Phase 3 to investing in a drug development...
Read article
May 20, 2015
Seamless adaptive clinical trials have gained popularity for reducing the projected time it takes to complete the...
Read article
May 15, 2015
Imagine that it’s been three years since the completion of a trial, and that suddenly a regulatory body calls into...
Read article
May 14, 2015
Ten years ago, in May 2005, world-renowned biostatistician Marvin Zelen was asked to deliver a keynote address before...
Read article
April 30, 2015
Adaptive financing (not to be confused with adaptive licensing) explores how biotechs, pharmaceuticals and potential...
Read article
April 23, 2015
Last week the Cytel Blog discussed the benefits of using the Adaptive Maximizing Design [AM Design] for dose-finding...
Read article
April 21, 2015
“We shouldn’t use an adaptive design, our sample size is too small.” Most clinical trial planners have heard this line...
Read article
April 16, 2015
When testing certain types of new drugs it is known in advance that the adverse side-effects of the medication will...
Read article
April 13, 2015
Cytel has published a new whitepaper on Monte Carlo Simulations for Patient Recruitment, which illustrates how a...
Read article
April 9, 2015
Last year. Nature Reviews Drug Discovery asked the FDA’s Tatiana Prowell (Hematology & Oncology Products Division)...
Read article
April 2, 2015
Midway through a trial is a terrible time to realize that you need a new strategy to complete the study. Sadly, it is...
Read article
March 27, 2015
We have often said that one of the greatest benefits of an adaptive clinical trial is the flexibility it affords for...
Read article
March 26, 2015
Last year Sunesis completed the VALOR trial, the first clinical study to make use of the groundbreaking promising zone...
Read article
March 20, 2015
Sometimes a new candidate drug for a pediatric study has already been tested on adults for safety and efficacy. We know...
Read article
March 19, 2015
A well-timed interim analysis can generally supply added benefits to the operational and administrative aspects of a...
Read article
March 12, 2015
Recently, we published an interview with Chris Conklin, the Director of the Center for Feasibility Excellence at...
Read article
March 10, 2015
Most of us are primed to think about the design of adaptive clinical trials as a narrow set of techniques applied to a...
Read article
March 5, 2015
The Mehta-Pocock promising zone is often used to carry out unblinded sample size re-estimation during interim analysis....
Read article
February 26, 2015
Cytel President and co-founder Cyrus Mehta has co-authored a paper on Infantile Hemangioma, recently published in the...
Read article
February 17, 2015
21st Century Cures (also called Cures2015) is a bipartisan initiative undertaken by the Committee on Energy and...
Read article
February 12, 2015
Data driven decision-making can ensure that every feasibility team achieves its enrollment milestones. By transforming...
Read article
February 10, 2015
Cardiovascular outcome trials (CVOTs) have earned the reputation of being the untamable behemoths of the clinical...
Read article
February 5, 2015
Every clinical trial requires some manner of trial forecasting, normally for feasibility and patient enrollment....
Read article
February 3, 2015
A new publication co-authored by Cytel Co-Founder and President Cyrus Mehta considers a range of clinical development...
Read article
January 29, 2015
February 2015 marks the five year anniversary of the FDA’s Guidance on Adaptive Design Clinical Trials for Drug and...
Read article
January 27, 2015
Last week we released an infographic on why Phase 3 trials fail. The numbers, while eye-opening, did not capture a...
Read article
January 22, 2015
The Journal of the American Medical Association recently published an article entitled ‘The Anatomy of Medical...
Read article
January 20, 2015
According to a recent Cytel Whitepaper on Adaptive Clinical Trials, 50% of Phase 3 trials eventually fail. This new...
Read article
January 16, 2015
Adaptive designs are thought to be the new paradigm in drug development, allowing statisticians and trial designers to...
Read article
January 13, 2015
Janus was the Roman God of transitions, a deity with two faces, one looking towards the past and the other the future....
Read article
December 18, 2014
Earlier this week, we at Cytel enjoyed a riveting in-house discussion on the uses of Bayesian decision rules for...
Read article
December 16, 2014
Richard Branson once wrote: “I have always valued capability over expertise. While you may need to hire specialists for...
Read article
December 11, 2014
A common framework for the clinical development of vaccines involves the study of several candidate compounds in Phase...
Read article
December 9, 2014
When planning a conventional trial, one can anticipate the drug supply necessary for the trial by determining how the...
Read article
December 4, 2014
Fulyzaq® from Napo/Salix was the first drug ever to be approved using an adaptive two-stage "seamless" clinical trial...
Read article
December 2, 2014
During a recent DIA webinar on reinventing the clinical trial, Laurie Halloran (President of the Halloran Consulting...
Read article
November 18, 2014
Statisticians and scientists at Novartis have been at the forefront of developing a new method in early phase oncology...
Read article
November 17, 2014
Outstanding Thought Leader, Scientist and Humanitarian, Passes away at Age 87 It is with immense sadness that we...
Read article
November 12, 2014
Clinical utility functions provide Phase 2 trial sponsors with an intuitive metric by which to measure the quality of a...
Read article
November 6, 2014
Professor LJ Wei holds that rules are for lawyers, not (necessarily) clinicians. When designing modern clinical trials,...
Read article
November 4, 2014
A key stage of exploratory drug development is implementing a proof-of-concept study to demonstrate the safety of a...
Read article
October 30, 2014
During last week’s East Users Group Meeting, Michael Proschan of the NIH and NIAID, gave a presentation on ‘Blinded...
Read article
October 28, 2014
Phase 1 oncology trials typically use either rule-based methods or model-based methods to determine the most acceptable...
Read article
October 21, 2014
Every year, the East Users Group Meeting brings together notable experts from industry and academia to discuss the...
Read article
October 14, 2014
We are often asked how statistical consultants can add value to the clinical development process. What do they...
Read article
October 7, 2014
Guest blogger Laurie Halloran is the President and CEO of Halloran Consulting Group, a management consulting firm for...
Read article
October 2, 2014
Sofia S. Villar is a member of the DART (Design and Analysis of Randomised Trials) group at the MRC Biostatistics Unit...
Read article
September 25, 2014
A few weeks ago, we posted a synopsis of an event held at ISCB Vienna in which statisticians from Cytel, SAS and Stata...
Read article
September 23, 2014
If you're a company considering an adaptive trial for the first time, you probably have many questions. One of our...
Read article
September 18, 2014
When designing clinical trials, many trial designers are advised to keep the trial simple. Prima facie, the keep it...
Read article
September 11, 2014
According to a recent Impact Report from the Tufts Center for the Study of Drug Development, 21% of active clinical...
Read article
September 4, 2014
Imagine if we were to count the number of possible reasons that investigators might have for monitoring a biomarker...
Read article
September 2, 2014
As more clinical trials make use of adaptive designs, investors have come to realize that high quality trial designs...
Read article
August 26, 2014
During an invited speakers session at the lnternational Society for Clinical Biostatistics, Cytel VP Yannis Jemiai was...
Read article
August 21, 2014
Use of the continual reassessment method (CRM) is safe, efficient, and flexible, according to a comprehensive review of...
Read article
August 14, 2014
Cytel statisticians are looking foward to attending the Conference of the International Society for Clinical...
Read article
August 12, 2014
Beyond Wild Horses: Developing Innovation at Cytel "Horse-and-pony" by arjecahn on flickr. -...
Read article
August 5, 2014
Cytel CTO Nitin Patel, recently sat down with ECHOES (a magazine for statistics in clinical trials) to discuss his...
Read article
July 31, 2014
Statisticians at Cytel are staunch advocates of the use of Bayesian methods in clinical trials. This summer's Joint...
Read article
July 29, 2014
Adaptive designs are the unsurprising hot topic of this year’s Joint Statistical Meeting, which features over one...
Read article
July 24, 2014
This is the third post in a three part series in which we consider (i) improvements to trial quality that result from...
Read article
July 22, 2014
PROCYSBI, the first drug to receive FDA approval after following an adaptive population re-assessment design, was one...
Read article
July 17, 2014
In anticipation of Cyrus Mehta’s webinar next week on new predictive analytics tools for trial forecasting, we thought...
Read article
July 15, 2014
Do you design or analyze Phase 1 dose escalation trials? Have you considered methods other than 3+3? This new Cytel...
Read article
July 10, 2014
Complexities with identifying suitable test populations in oncology studies contribute significantly to the 60%...
Read article
July 8, 2014
The above graphic is from Cyrus Mehta's slides on 'Adaptive Population Enrichment for Oncology Trials with Time to...
Read article
July 1, 2014
A recent Cytel Seminar on Adaptive Statistical Designs featured a talk by Michael Elashoff (Patient Profiles) on...
Read article
June 26, 2014
The rise of biomarker based treatments in oncology has meant a reconceptualization of what constitutes a particular...
Read article
June 10, 2014
The FDA’s Tatiana Prowell (Breast Cancer Scientific Lead in the Office of Hematology & Oncology Products) recently gave...
Read article
June 5, 2014
Charles Liu, PhD, Cytel Statistician and Product Manager In the US, cancer is the most common cause of death after...
Read article
June 3, 2014
This is the second post in a three part series in which we consider (i) improvements to trial quality that result from...
Read article
May 29, 2014
Cytel has taken the initiative to train the next generation of clinical programmers through its innovative Clinnical...
Read article
May 22, 2014
This is the first of a three part post in which we will consider (i) improvements to trial quality that result from...
Read article
May 19, 2014
Pantelis Vlachos, PhD, is a Director at Cytel Consulting. He works with a team of experts who regularly assist clinical...
Read article
May 13, 2014
The VALOR trial recently applied a promising zone design to a Phase 3 evaluation of Vosaroxin, a candidate for the...
Read article
May 8, 2014
( Editor's note: This post has been refreshed in December 2016) Model based algorithms for Phase I dose-escalation have...
Read article
May 5, 2014
A new JAMA study on discontinued randomized trials in Switzerland, Germany and Canada, reports that poor recruitment...
Read article
April 30, 2014
For the second installment of our StatXact 25th Anniversary Retrospective Series, Professor Joan Hilton (UC San...
Read article
April 24, 2014
Here at Cytel we firmly believe that if you don’t get the design of a clinical program right, then nothing else...
Read article
April 21, 2014
The core methodological problem that would eventually spur the development of Cytel’s StatXact software was first posed...
Read article