

At the November PhUSE-EU Connect in Frankfurt, I presented a poster entitled "The « CDISC Stupidario » (the CDISC Nonsense)." In the poster and in the paper presented during the event I wanted to share the main CDISC “Nonsense” I have seen in my daily activity as a CDISC SME in reviewing both internal (Cytel) and external CDISC packages (delivered by Pharma or other CROs). Examples can range from "nonsense" questions to a complete misunderstanding of the CDISC Ig(s). Ultimately the goal of my presentation was to raise the importance of the correct use of standards and to highlight that details really do matter.
While in the paper I described the CDISC “nonsense” in detail, in the poster session I invited the PhUSE attendees to try to find the issue themselves and the reason why the different real scenarios described in the poster should be considered a “Stupidario."
The poster session on the inaugural day of the conference was quite intensive, and many of the attendees accepted the challenge and spent some time trying to answer my ‘quiz.' It was very encouraging to see that although the level of the expertise of the attendees I interacted with ranged a lot, from people with limited CDISC and submission experience to people with knowledge of one standard but not the other, the majority of my quiz was correctly answered by many different attendees.
If you didn’t attend the poster presentation, why not challenge yourself now? Access the poster via the button below and test yourself before reading the rest of the blog below for the answers to the ‘fill the gaps’ or determine what is wrong with the proposed scenarios.
Click the button to review the poster and test yourself before reading the answers below!

1. "Too lazy to correct issues."
Very often people pay different "attention" to details, perhaps (incorrectly) thinking they do not matter. Let's say for example that you created an SDTM dataset, and for one variable you assigned the wrong label (not as per the SDTM Ig), or you did not assign a label at all. When the Pinnacle21 validation outcome detects this, why not correct the issue in the dataset instead of leaving the issue as it is and justifying it in the study data reviewer guide as a "Programmatic Error"?
2. "Where is my traceability?"
Traceability in ADaM Ig is continuously “stressed” making this principle key for a good ADaM dataset. One aspect stressed in the ADaM Ig is the transparency that you provide by making your ADaM fully traceable:
Retaining in one dataset all of the observed and derived rows for the analysis parameter provides the clearest traceability most flexibly within the standard BDS. The resulting dataset also offers the most flexibility for testing the robustness of an analysis.
So unless you end up with a huge ADaM dataset, keep the original source records in it: this will beneficial for the traceability of your ADaM derived variables, and ultimately it will be transparent for the reviewer which records have been used (and eventually which records have been not used or not analyzed).
3. "My PARAM should be enough."
Why do we need AVALU? AVALU is what I call a "decorative variable" since PARAM should already contain this information; from ADaM Ig PARAM must include all descriptive and qualifying information relevant to the analysis purpose of the parameter (ADAM Ig 1.1 Section 3.3.4), e.g. unit PARAM=" Weight (kg)."
The other common "mistake," although not really a conformance issue, is to create PARAM by concatenating, for example, all SDTM findings qualifiers of the –TEST variable, for example for ECG "Systolic Blood Pressure (mmHg), Sitting Position." This is not really needed unless you are doing an ECG analysis based on the subject position. Again this is clear from the ADaM Ig section 3.3.4: PARAM must include all descriptive and qualifying information relevant to the analysis purpose of the parameter, this means, in few words, that in most of the cases concatenating the parameter name and its unit should be enough (this is what usually goes into the statistical output).
4. “ADaM ‘Superfluo’ Submission”
How many analysis ADaM datasets do I need to create given the fact my study SDTM has 22 subject datasets?
There is, of course, no answer without knowing the details of the statistical analysis to be performed. ADaM, in contrast to SDTM, is analysis-driven, meaning that you do not need to create an ADaM dataset for laboratory data if your Statistical Analysis Plan does not plan for any aggregate analysis on laboratory data. For example, you might create listings directly from your SDTM if you don't need any major derivations, and very likely you will not need an ADIE ADaM dataset. ADIE what? An ADaM dataset for violated Inclusion and Exclusion Criteria. This is really not needed!
These are some of the recommendations we give on this topic during the official CDISC ADaM training:
What analysis datasets are required for a submission?
ADaM ADSL dataset is required. FDA expects additional ADaM datasets to support primary and secondary analysis. From the FDA Technical Conformance Guidance: Sponsors should submit ADaM dataset to support key efficacy and safety analysis and the primary and secondary endpoints of a trial should be provided as well.
Should there be an ADaM dataset for every SDTM domain?
No. There is no requirement that every SDTM domain has a corresponding analysis dataset. ADaM datasets are needed to support the Statistical Analysis Plan, and they are not merely created as a mechanistic duplication of SDTM data.
5. "Something is wrong with the terminology."
The wrong thinking here was that the version of the SDTM Ig had to “parallel” the CDISC CT version, but there are actually no requirements to have Ig and CDISC CT from the same period and therefore, despite working with SDTM Ig 3.1.3, a more recent version of the CDISC CT could have been implemented, e.g. one of the 2018 CDISC CT.
6. “Something is wrong with my aCRF bookmarks."
As of today there is no industry standard on how the SDTM Annotated CRF (aCRF) should be created, but the CDISC "Metadata Submission Guideline (MSG) for SDTM IG" is a valid document that sponsors and CROs should refer to with regards to aCRFs, in particular, section 4 "Guideline for Annotating and Bookmarking and CRFs” .
From the guidance:
Annotated CRFs included in the eCTD should be bookmarked two ways (dual bookmarking): bookmarks by time-points, often analogous to planned visits in the study, and bookmarks by CRF topics or forms. SDTM domains do not necessarily have a 1-to-1 relationship with CRF topics or forms, nor is the reverse true. For example, in the annotated CRF, both DM and SC are collected on the Demography panel, while SC data are collected from the Enrolment Form and the Demography pages
The following figures show the wrong interpretation of the above sentence (left side) and the correct one (right side)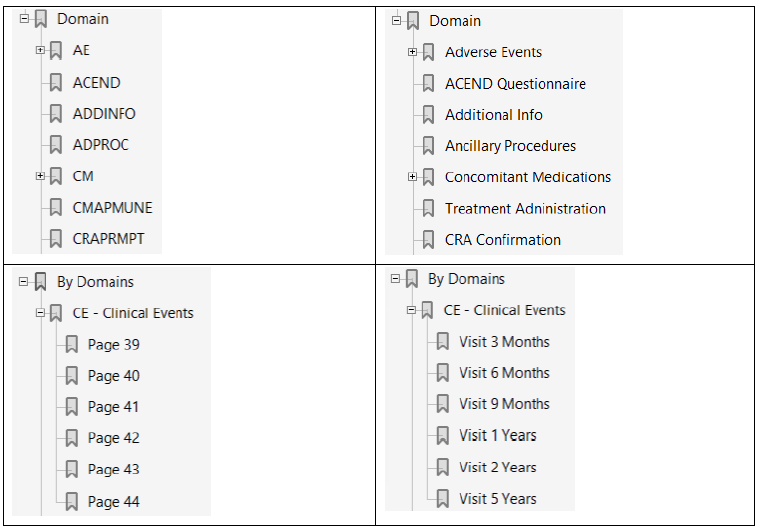
See also here from the recent PhUSE event an additional proposal from the CDISC Germany User Network.
7. What standard is CDISC for?
CDISC is only for data standards. There are no industry standard output templates yet. The PhUSE initiative has released some white papers with some recommendations; however, the examples provided in these white papers do not yet represent an industry standard and companies are still free to follow their organizational standards when developing statistical outputs (tables, listings, and figures). Once again, this is not officially connected with CDISC ….. or at least as of today.
8. “I’m your reviewer, can you please say something more in the data conformance summary section of your Clinical Study Data Reviewer Guide (cSDRG)?”
The conformance summary section of the cSDRG should not be a simple list of issues detected by the validator tool and a clear rationale “justifying” the unresolved issues should be provided. "Resolvable" issues should be addressed, so they are no longer issues. Believe it or not, reviewers pay attention to such details, and on more than one occasion we have seen comments back from for example the FDA reviewer asking for more information or a better-explained rationale.
The examples reported in the poster could have been better explained, and additional rationale could have been provided as follows:
1. Although –DY variable is permissible and sponsor could omit it, the FDA Study Technical Conformance Guide requires –DY variable to be included when –DTC is included in the data. Merely saying "Variable not used" does not matter!
2. This is an incorrect implementation! --TEST should have been abbreviated and full text specified in the cSDRG.
3. Does it mean the sex was collected, but for some subjects, the information was not available in the original data?
4. Incomplete coding might be the subject of rejection from the PMDA and a major concern of the FDA. You need a strong rationale, and therefore the explanation should have provided the reason why the term was not coded.
5. More details about laboratory parameters and units concerned should have been mentioned.
In conclusion, the efficacy and safety of your drug are of course ultimately what matter, but lack of traceability, inadequate or insufficient documentation might trigger questions and concerns from the reviewer. You may think these are minor issues because they do not ultimately impact any results. However, you are risking your credibility with the FDA reviewer, who may conclude that your package is not of sufficient quality.
Would you like to discuss these elements in more detail and learn more? Join me and the Cytel team at the next CDISC EU Interchange in Amsterdam from May 6th to May 7th, where you have several options to connect and discuss your next CDISC project:
Attend any of the two CDISC ADaM trainings planned on the May 6th-10th in Amsterdam.
Attend my presentation on May 8th in the afternoon, where I will show more ‘Stupidario(s)’
Join the Foundational Stream which I’m chairing on May 9th
Visit my Poster “A Systematic Review of CDISC Therapeutic Area User Guides - TAUGs”
Stop at Cytel booth any time you like on May 7th or 8th
To arrange a meeting at the CDISC Interchange click the button below:
