

The Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER) are the two main divisions of the FDA responsible for regulating drugs and biological products, as well as combinations of drugs and biological products. CDER focuses on drug regulation, whereas CBER oversees the regulation of biological products.1
CDER and CBER share many common principles and processes when evaluating products for market approval, including process and guidance requirements for data standards submission.2 However, submitting to the CBER division is unique and has additional requirements compared to other divisions, not only when submitting data packages in support of new vaccine approvals (Office of Vaccines Research and Review, OVRR),3 but with all submissions requesting data submission details in a dedicated Study Data Standardization Plan (SDSP), such as the structure of your datasets.
The “Vaccines Technical Specification Guidance,” initially released in April 2018 and followed by a second version in December 2019, was introduced during an FDA webinar.4 Over the years, several sponsors have shared their experiences and the impact of this guidance on various aspects of their processes, including data governance, medical monitoring, SOPs, data entry guidelines, and Case Report Form (CRF) design.5,6,7 The goal is to provide CBER reviewers with the background needed to understand the purpose of the data and, eventually, to offer recommendations. This could involve remapping data to avoid custom domains or unnecessary mapping to supplemental qualifiers. For example:
- For safety: reactogenicity (CE, FACE, and VS), unsolicited adverse events (AE), including medical attended AEs, death (AE, DM, and DD), laboratory safety assessment (LB).
- For efficacy: clinical disease endpoint (CE and MB) and immunogenicity (IS).
CBER places a strong emphasis on early discussions with sponsors during the drug development process. These discussions encompass recommendations not only on how data should be standardized and submitted but also how data should be collected. Therefore, alongside the SDSP, CBER requests the submission of the SDTM aCRF.
Experiencing the CBER
At Cytel, our programming and statistical team has accumulated extensive experience in submitting data and interacting with health authorities worldwide, notably the FDA. This experience includes working on several Integrated Summaries of Safety (ISS) and Integrated Summaries of Effectiveness (ISE).8,9,10,11 Among the experiences we had submitting data packages to CBER, a clear cut was between the ones we did just around the release of CBER and the ones after the guidance was released (Figure 1).
Figure 1: Two CBER submissions before and after CBER guidance / webinar
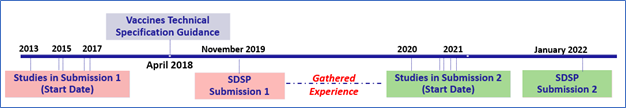
Figure 2 and Figure 3 summarize the flow of the communication with the CBER reviewer, the kind of requests we got and how we addressed them, whether we were able to implement them all or not.
Figure 2: A non-vaccine submission to CBER

Figure 3: A vaccine submission to CBER

For the second submission in particular, the experience gained from the previous submission was invaluable, as it allowed the new team to receive relevant training and conduct a gap analysis to align with CBER’s requirements. Like the previous experience, the appointed CRO was able to address some of our requests, but not all.
The submission process was akin to the previous experience, involving the provision of a SDSP that included a specific CBER appendix. This time, we also had to anticipate potential non-conformance issues. As an outcome, in the second submission, we did receive half of the request to change compared to previous submission, 19 vs 10 main items to correct, of which about one-third were not applicable.
In both cases, after team discussions, we compiled our responses into a single document and incorporated them into a new version of the SDSP, which we subsequently re-submitted to the FDA CBER.
Conclusions
Submitting data packages to CBER introduces distinct challenges, even for sponsors experienced in implementing CDISC standards throughout their product development. CBER’s requirements add an additional layer of complexity on top of those already published by CDISC, notably the SDTM Implementation Guidance.
As emphasized in other public sponsor discussions, “at this time, the CBER guidance itself as well as the Vaccine and COVID-19 CDISC TAUGs are out-of-date. Sponsors must be able to adapt quickly while maintaining quality and integrity of the clinical trial data and make informed decisions for ongoing and completed studies; especially if the changes are not quick or easy to implement.”
It's important to note that when studies have already commenced, and data collection is in progress or even completed, some of CBER’s recommendations may not be easily implementable. This is often the case, as most of the studies in our submissions were either ongoing or already completed. For instance, recommendations to include checkboxes for indicating reactogenicity events during a prespecified time frame or checkboxes to indicate daily event collection during the assessment interval may not be feasible if the eCRF has already been designed and validated for deployment.
For this reason, it is important to start discussions with CBER division very early during the drug development process. These discussions encompass recommendations not only on how data should be standardized and submitted but also how data should be collected. Therefore, alongside the SDSP, CBER requests the submission of the SDTM aCRF.
I will be presenting “Experiencing the CBER” at PHUSE EU Connect in Birmingham, November 5–8, 2023. Stay tuned (and subscribe to our blog) for more information on this upcoming event!
Will you be attending PHUSE EU on Nov. 5–8? Click below to book a meeting with our experts:

Notes
1. T. White, “FDA Overview – CDER vs. CBER.”
2. A. Tinazzi, “Raising Awareness for Additional FDA Data Standards Submission Recommendations,” CDISC-EU Interchange, 2023 (copy of slides and paper available upon request).
3. FDA CBER, “Vaccines Technical Specification Guidance,” v2.1, Dec 2019.
4. “Optimizing Your Study Data Submissions to FDA: Office of Vaccines Research and Review (OVRR) Data Submission,” May 8, 2018.
5. S. VanPelt Nguyen, E. Bernstein-Hanlon, L. Houterloot, R. Li, “Challenges Addressing CBER Guidance on Reactogenicity, from Collection to Analysis,” PHUSE US, 2023.
6. S. VanPelt Nguyen and L. Zhang, “Challenges with Implementation of New Standards and Guidance – A Sponsor’s Experience with the April 2018 CBER Vaccine Guidance,” PHUSE US, 2023.
7. J. Mastri and S. McLaughlin, “Adapting and Evolving with OVRR Requirements,” CDISC-EU, 2023.
8. Florence Le Maulf, “Presenting Clinical Data for Regulatory Submission: A Stats Perspective,” Cytel Blog.
9. F. Le Maulf, “Introduction to ISS/ISE,” IX Italian CDISC User Network, 2023.
10. A. Tinazzi, “The Integration Dilemma,” PHUSE-EU, 2022.
11. “The Good Data Doctor on Data Submission and Data Integration,” Cytel ebook, 2023.
Read more from Perspectives on Enquiry and Evidence:
Sorry no results please clear the filters and try again

Preparing Your Integrated Summaries of Safety and Effectiveness: Best Practices

Presenting Clinical Data for Regulatory Submission: A Stats Perspective

New Ebook: “The Good Data Doctor on Data Submission and Data Integration”