April 10, 2024
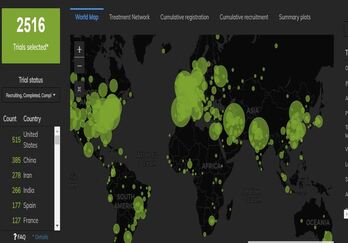
Orphan drug designation is a regulatory status granted to pharmaceuticals developed for the treatment of rare diseases....
Read article
December 20, 2023
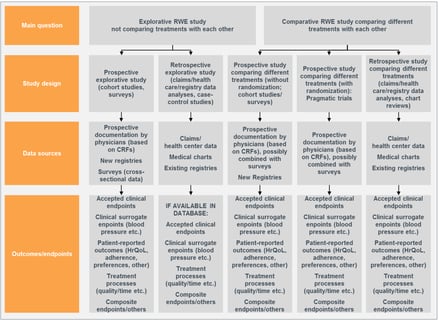
Perspectives covers a wide range of topics related to real-world evidence and real-world data, from overcoming health...
Read article
December 15, 2023
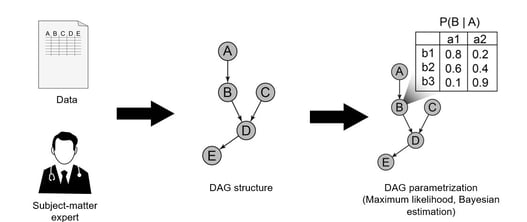
In clinical trials, high-quality data is essential. It drives the drug development decision-making process and is a...
Read article
December 8, 2023
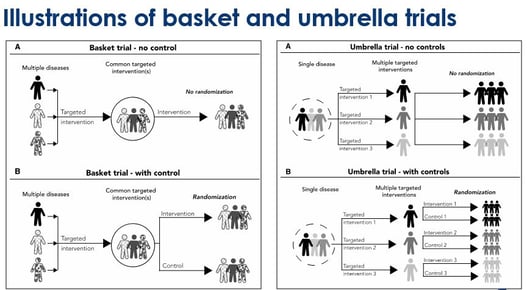
Regular technical discussions with the FDA play a critical role in ensuring data submission success. These discussions...
Read article
October 18, 2023
Written by Angelo Tinazzi and Florence Le Maulf Integrated Summaries of Safety (ISS) and Integrated Summaries of...
Read article
August 25, 2023
For several years, CDISC and Regulatory Data Submission expert Angelo Tinazzi has authored the series, The Good Data...
Read article
June 15, 2022
For years CDISC data standards implementers have struggled to find good implementation examples and use cases beside...
Read article
July 30, 2021
I had recently (for the first time) the pleasure and honor to attend a virtual meeting with the FDA, a pre-NDA Type-B...
Read article
April 13, 2021
There has been an increasing use of digital measures in drug development recently. New wearables technologies can help...
Read article
March 30, 2021
As of today, our Industry has not defined any approach, nor does an official regulatory agency...
Read article
February 25, 2021
The convergence of several distinct trends has made wearables an increasingly attractive option for use in confirmatory...
Read article
February 24, 2021
In a previous post, I discussed the importance of proper use of CDISC Controlled Terminology (CDISC CT) in SDTM....
Read article
February 11, 2021
The past two years have witnessed a heightened interest in the use of wearables in clinical development. The unexpected...
Read article
December 21, 2020
2020 has been an unusually difficult year as the global pandemic impacted all of our lives. This year, the Cytel blog...
Read article
December 18, 2020
Can I submit software programs other than SAS? What software programs should I submit? Are sponsors required to submit...
Read article
December 1, 2020
An extraordinary amount of global research is underway as the COVID-19 pandemic continues to evolve and spread. As...
Read article
November 17, 2020
As a part of Cytel’s "New Horizons Webinar Series", Alind Gupta, Senior Data Scientist, presents case studies from his...
Read article
October 13, 2020
The combination of greater access to electronic health records, bigger electronic claims datasets, and the need for...
Read article
October 12, 2020
Cytel and Novartis are together hosting a complimentary Bayesian Virtual Symposium and an Interactive 7-part workshop....
Read article
September 14, 2020
“A good start is half the battle” (the Before) when submitting data to the FDA and there are a couple of cherries to...
Read article
September 4, 2020
On Friday September 11, Cyrus Mehta, co-founder of Cytel, will be delivering a talk to the Heart Failure Collaboratory,...
Read article
July 28, 2020
CDISC standards have been around for a while with the first SDTM Standard version released in 2004. However, it was...
Read article
June 29, 2020
From the time the COVID-19 outbreak was declared a pandemic, the number of studies conducted around the world to either...
Read article
April 29, 2020
In our previous blog, we spoke with Alind Gupta, who works as a Machine Learning Researcher at Cytel in Canada. The...
Read article
April 27, 2020
In the first part of this two-parts blog, I speak about how the European CDISC Committee (E3C) together with CDISC...
Read article
April 24, 2020
In early March, when countries around the world started implementing lockdowns, the European CDISC Committee (E3C)...
Read article
April 20, 2020
Cytel is hosting a webinar on Transparent Machine Learning in Oncology, on April 21, 2020. Our speaker, Alind Gupta,...
Read article
March 31, 2020
Since 1953, when the discovery of the structure of DNA was made, we have seen great advancements in genomics....
Read article
October 1, 2018
One of my wife’s favorite TV shows is ‘Quattro Ristoranti’ (Four Restaurants). In each episode of the show, 4...
Read article
September 7, 2018
On August 29th 2018, the FDA announced (1) that it would be establishing a Complex Innovative Trial Design (CID) Pilot...
Read article
October 27, 2017
In this blog, we share the second part of our interview with Bob Beckman, about a design concept for a confirmatory...
Read article
May 12, 2016
Once upon a time Hansel and Gretel laid a trail of breadcrumbs which they followed to find their way back home. Their...
Read article
April 13, 2016
We continue our series of blogs covering the expert presentations from the EAST User Group Meeting. Consultant Claire...
Read article
May 15, 2015
Imagine that it’s been three years since the completion of a trial, and that suddenly a regulatory body calls into...
Read article
June 10, 2014
The FDA’s Tatiana Prowell (Breast Cancer Scientific Lead in the Office of Hematology & Oncology Products) recently gave...
Read article